
The TNNI3 p.R186Q mutation is responsible for hypertrophic cardiomyopathy via promoting FASN-stimulated abnormal fatty acid metabolism
 0
0
Abstract
Introduction: The TNNI3 gene encodes the protein of cardiac troponin I (cTnI), which is an inhibitory subunit of sarcomeres. Mutations in this gene account for 3% of hypertrophic cardiomyopathy (HCM) and the molecular mechanism is complex. Recently, lipid metabolism has been revealed to be involved in HCM.
Aim: The purpose of this work is to identify whether the pathological mechanism of the hotspot mutation TNNI3 p.R186Q in HCM is related to abnormal lipid metabolism.
Methods and Results: A knock-in (KI) mouse model carrying the Tnni3 p.R186Q homozygous mutation
Conclusion: In the present study, we successfully engineered Tnni3R186Q/R186Q mice with the typical phenotype of myocardial hypertrophy. We demonstrated that the TNNI3 p.R186Q mutation could induce HCM by the dissociation of EGFR and cTnI, which further led to EGFR-dependent increased expression of FASN and abnormal lipid metabolism.
Keywords
INTRODUCTION
Hypertrophic cardiomyopathy (HCM), characterized by asymmetrical hypertrophy of the left ventricle and ventricular septum, is a familial autosomal dominant hereditary cardiomyopathy[1,2]. Generally, there are five common pathogenic genes: β-myosin heavy chain (MYH7), cardiac myosin binding protein C3 (MYBPC3), Troponin T2 (TNNT2), Troponin I3 (TNNI3) and Tropomyosin 1 (TPM1)[3]. Among them, the TNNI3 gene encoding the protein of cardiac troponin I (cTnI) protein is an inhibitory subunit of the sarcomeres. To date, more than 55 mutations of TNNI3 have been linked to cardiomyopathy and account for approximately 3% of HCM[4]. Several studies have also reported that the TNNI3 p.R186Q mutation is associated with HCM and a high risk of sudden death[4,5]. Similarly, we previously discovered the TNNI3 p.R186Q mutation in a 39-year-old proband from an HCM family of Han Chinese individuals[6]. Nevertheless, to date, the molecular mechanisms responsible for the HCM pathogenesis of TNNI3 p.R186Q, which is a hotspot, are poorly understood.
It is well known that much of the ATP necessary for heart contraction mainly depends on the oxidation of fatty acids (FAs)[7], and oxidation and synthesis of FAs maintain the dynamic balance of FAs under physiological conditions. However, the imbalance of FAs can result in many different cardiovascular diseases. Previous studies have found that increased fatty acid synthesis leads to lipid accumulation and lipo-toxicity by disrupting FAs balance, eventually developing into HCM[8,9]. Fatty acid synthase (FASN), a key metabolic multi-enzyme, is responsible for the de novo synthesis of long-chain FAs, which play an essential role in fatty acid metabolism. Studies have shown that the overexpression of FASN is related to the occurrence, evolution, invasion and prognosis of tumors[10,11]. Nevertheless, until now, the influence of FASN on the heart remains unclear. Strikingly, our preliminary work showed that the expression of FASN was significantly enhanced in the hearts of mice carrying the Tnni3-R186Q mutant allele (Tnni3R186Q/R186Q). Thus, the purpose of this study was to further explore the influence of FASN on TNNI3-R186Q in HCM.
MATERIALS AND METHODS
Animals
Neonatal SD rats at 3 days were purchased from Shanghai Laboratory Animal Centre (Shanghai, CAS). C57BL/6J Knock-in (KI) mice were engineered through homologous recombination with help from Shanghai Biomodel Organism Science & Technology Development. DNA sequencing confirmed the successful construction of KI mice and the germline transmission of the TNNI3 p.R186Q point mutation. Standard rodent sterile chow and water were given ad libitum in individually ventilated cage systems (IVC systems). All animal care procedures and experiments were approved by the Animal Ethics and Experimentation Committee of Nanchang University and conformed to the “Guide for the Care and Use of Laboratory Animals” (revised 1996). According to the genotype, the experimental KI mice were divided into Tnni3R186Q/R186Q and Tnni3R186Q/+ mice and the littermates of Tnni3+/+ mice were matched by age and sex. Adult mice of both genders were used in the present study. There were no significant differences in the littering period, litter size, growth and behavior of the Tnni3R186Q/R186Q, Tnni3R186Q/+ - and Tnni3+/+ mice, respectively.
TNNI3 protein interaction network analysis
A protein-protein interaction (PPI) network of TNNI3 was constructed using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database Available from: https://www.string-db.org/.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS)
Proteins were extracted from Tnni3R186Q/R186Q and Tnni3+/+ mice heart tissues and analyzed by label-free tandem mass tag (TMT) and LC-MS/MS. The KEGG analysis was done with the help of Shanghai Applied Protein Technology Co., Ltd., who provided technological assistance.
Cell preparation and plasmid construction
The neonatal rat cardiomyocytes (NRCMs) used in the experiments were derived from newborn rats within three days, according to a previously published protocol[12]. The adenoviral vectors (adenovirus TNNI3 p.R186Q mutation, Ad-Mut; adenovirus wild type, Ad-WT) were obtained from Invitrogen. All cells were seeded into 6-well plates 1 day prior to transfection at a density of 1 × 105 cells/well. The cells were infected with adenovirus at an MOI of 50 and re-incubated with DMEM supplemented with 10% FBS 8 h following transfection. After 48 h, the transfection effects were confirmed by fluorescence microscopy and western blot analysis. Plasmids and reagents are described in the Supplementary Materials and Methods.
Doppler echocardiography and histopathological analysis
Transthoracic echocardiography was performed as previously described[13] (Vevo 2100; Visual Sonics, Canada). Wheat germ agglutinin-fluorescein isothiocyanate-staining (WGA), hematoxylin and eosin (HE) and Masson’s trichrome staining were performed as previously described[13,14]. Please refer to the expanded materials and methods in the supplement for details.
Tandem mass tag-based quantitative proteomic analysis
Tandem mass tag-based quantitative proteomic analysis was performed at Shanghai Applied Protein Technology Co., Ltd.
Statistical analysis
All experimental results and experimental data were statistically analyzed and processed by SPSS 17.0 software. Continuous data were presented as the means ± standard error of the mean (SEM). Comparison data between the two groups were performed using unpaired Student’s t-tests (equal variance), or unpaired Student’s t-tests with Welch’s correction (unequal variance). Comparison data between multiple groups were performed using one-way analysis of variance (one-way ANOVA) with post hoc Tukey’s tests. GraphPad Prism 5.0 software was used to plot all statistical data. Statistical significance was defined as
RESULTS
The TNNI3 p.R186Q mutation facilitates cardiac hypertrophy in vivo and in vitro
The TNNI3 p. R168Q mutation found in a 39-year-old female HCM proband with a family history of sudden cardiac death was reported by the Hong group[6]. High pathogenicity possibility was predicted for the mutation by multiple bioinformatics tools [Supplementary Table 1].
To explore the effect of the TNNI3 p.R168Q mutation on myocytes in vivo, we engineered a KI mouse model harboring the mutation (Tnni3R186Q/R186Q, Tnni3R186Q/+) by CRISPR/Cas9 technology in point mutation in C57BL/6 mice [Supplementary Figure 1]. Compared with age- and sex-matched Tnni3+/+ littermates, the cTnI transcriptional and posttranslational expression levels were no different among the Tnni3+/+,
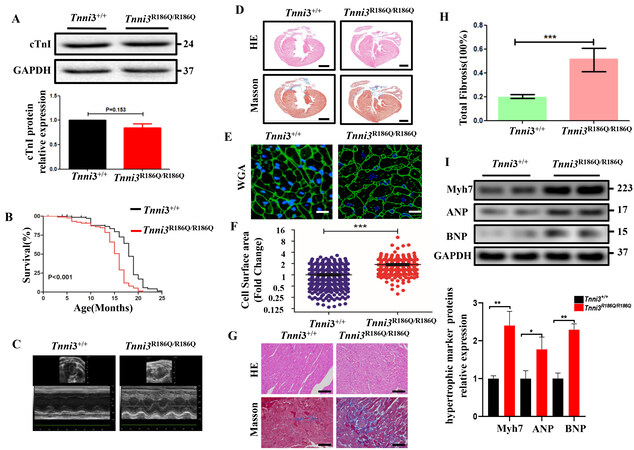
Figure 1. Tnni3R186Q/R186Q mice exhibit cardiomyocyte hypertrophy. (A) The cTnI protein expression level in sections of the ventricles of Tnni3+/+ and Tnni3R186Q/R186Q littermates (n = 3 per group). (B) The survival curve of Tnni3+/+ (n = 91) and Tnni3R186Q/R186Q (n = 78) mice. *P < 0.05, Log-Rank (Mantek-Cox) tests. (C) Representative M-mode echocardiographic images of Tnni3+/+ and Tnni3R186Q/R186Q mice at 10 months old. (D) Gross histological view (longitudinal section) of Tnni3+/+ and Tnni3R186Q/R186Q mice ventricular myocardium stained with HE (pink) and Masson’s trichrome staining (red) to reveal myocardial structure (100×). Scale bar 2 mm.
Parameters of heart function and structure detected by Echocardiography
| ECHO | Tnni3+/+ | Tnni3R186Q/R186Q |
| (n = 8) | (n = 8) | |
| LVIDd (mm) | 3.92 ± 0.26 | 3.27 ± 0.26*** |
| LVIDs (mm) | 2.46 ± 0.29 | 1.92 ± 0.40* |
| IVSd (mm) | 0.80 ± 0.06 | 1.02 ± 0.18* |
| IVSs (mm) | 1.28 ± 0.08 | 1.54 ± 0.13* |
| LVPWd (mm) | 0.86 ± 0.02 | 1.00 ± 0.13 |
| LVPWs (mm) | 1.18 ± 0.11 | 1.46 ± 0.16* |
| EF (%) | 67.91 ± 4.11 | 72.89 ± 9.19 |
| FS (%) | 37.39 ± 3.17 | 41.68 ± 8.34 |
In vitro, the adenovirus expressing the TNNI3 p.R168Q mutation (Ad-Mut) was transfected into NRCMs. Consistent with the above data in vivo, increased expression of hypertrophy markers was observed in the Ad-Mut group [Supplementary Figure 5]. Overall, these results suggested that the TNNI3 p. R168Q mutation in myocytes can induce the HCM phenotype.
Tnni3R186Q/R186Q mice exhibit aberrant fatty acid metabolism
To elucidate the mechanism of the TNNI3 p. R168Q mutation in HCM, the TMT coupled with LC-MS/MS was used to screen for differential proteins and the KEGG enrichment analysis was performed in Tnni3+/+ and Tnni3R186Q/R186Q mouse hearts at 10 months. The differentially expressed proteins from the KEGG pathways were mainly related to fatty acid metabolism [Figure 2A], which suggested that abnormal fatty acid metabolism might be involved in the pathogenesis of TNNI3 p.R186Q -related HCM. Consistent with this assumption, the results from Oil Red O assays showed that Tnni3R186Q/R186Q mouse hearts had a higher level of lip droplets [Figure 2B] Furthermore, through an enzyme-linked immunosorbent assay, the contents of total triglycerides (TG) and total cholesterol (TC) were increased in Tnni3R186Q/R186Q mouse hearts [Figure 2C and D], the content of the corresponding indicators in the serum did not increase
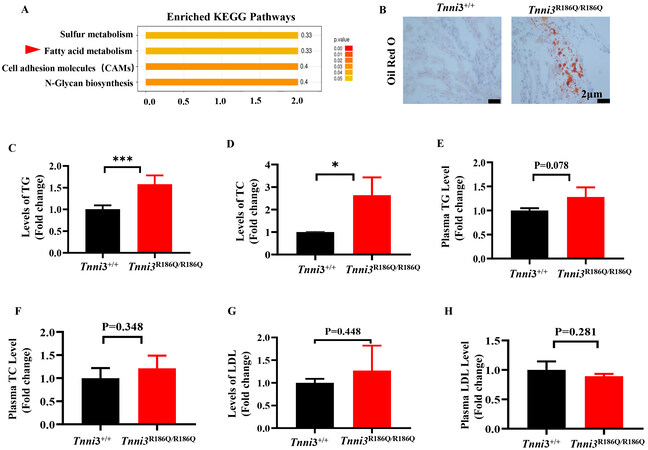
Figure 2. Tnni3R186Q/R186Q mice exhibit aberrant fatty acid metabolism. (A) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of differentially expressed genes in Tnni3+/+ and Tnni3R186Q/R186Q mice ventricular myocardium (n = 3 per group). (B) Lipogenesis in the ventricular myocardium from Tnni3+/+ and Tnni3R186Q/R186Q mice was determined by Oil Red O staining using frozen sections (n = 3 per group). (C-E) The levels of TG, TC and LDL were individually measured using enzyme-linked immunosorbent assay in the ventricular myocardium of Tnni3+/+ and Tnni3R186Q/R186Q mice (n = 3 per group). (F-H) The levels of TG, TC and LDL were individually measured using enzyme-linked immunosorbent assay in the serum of fasting Tnni3+/+ and Tnni3R186Q/R186Q mice (n = 3 per group).
The expression level of FASN is up-regulated in the hearts of Tnni3R186Q/R186Q mice
FASN is a key metabolic multi-enzyme, which plays an essential role in fatty acid metabolism. To explore the involvement of FASN in TNNI3R186Q/R186Q related fatty acid metabolism, both mRNA and protein expressions were determined in Tnni3R186Q/R186Q mice at 10 months. Consistent with our expectation, FASN expressions in both mRNA level and protein level were significantly increased in Tnni3R186Q/R186Q mice compared with those of Tnni3+/+ mice of the same age [Figure 3A-C]. As we know, the balance of fatty acid metabolism is jointly determined by fatty acid synthesis and beta-oxidation. Thus, we detected the expression of other related fatty acid synthesis (such as ACLY, SCD and PPAR-γ) and fatty acid β-oxidation genes (such as HADHB, ACAA2, ACOX-1, PPAR-a). However, we failed to observe any expression differences among these genes between Tnni3+/+ and Tnni3R186Q/R186Q mouse hearts [Figure 3D and E]. The above results suggested that the TNNI3 p.R168Q mutation leads to aberrant fatty acid metabolism by increasing the expression of FASN.
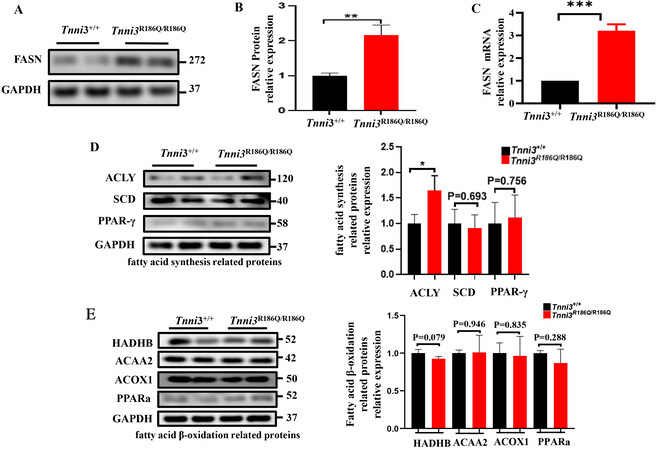
Figure 3. FASN expression is upregulated in TNNI3R186Q+/+ mouse hearts. (A) FASN protein levels measured by Western blot were detected in Tnni3+/+ and Tnni3R186Q/R186Q mice ventricular myocardium and a representative experiment is shown (n = 3 per group).
FASN inhibition attenuates TNNI3 p.R186Q-induced aberrant fatty acid metabolism and hypertrophy
To address whether TNNI3 p.R186Q-induced aberrant fatty acid metabolism and hypertrophy were dependent on FASN. Mice were treated with a suitable dose of C75[15], a FASN selective inhibitor by food intake for 2 months [Figure 4A]. Echocardiography detection showed that C75 did not affect cardiac structural changes [Figure 4B, Supplementary Table 3]. Furthermore, inconsistent with the above date, Oil Red O assays demonstrated that the increased lip droplets were profoundly reduced [Figure 4C], and the levels of TC and TG in mouse hearts were reduced [Figure 4D], and cardiac fibrosis [Figure 4E and F] and myocyte hypertrophy were attenuated [Figure 4G and H] by C75 administration in Tnni3R186Q/R186Q mice.
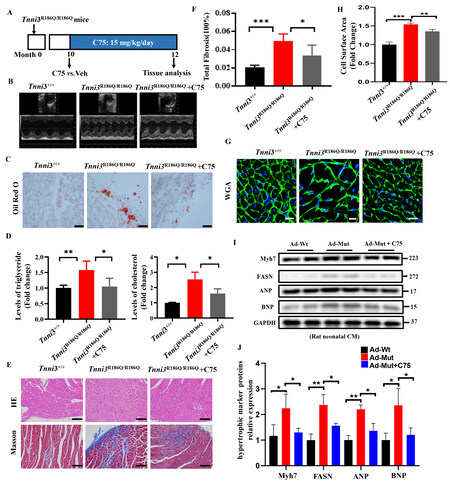
Figure 4. FASN inhibition attenuates TNNI3 p.R186Q-induced aberrant fatty acid metabolism and cardiac hypertrophy. Tnni3R186Q/R186Q mice at 10 months of age were administered saline or C75 (15 mg/kg/day) daily for 8 weeks. (A) Experiment timeline for
In vitro, when cells were treated with C75, the TNNI3 p. R168Q mutation-stimulated higher expression of hypertrophic marker was largely inhibited [Figure 4I and J]. Taken together, these results suggested that TNNI3 p.R186Q related HCM is dependent on FASN.
EGFR is essential for TNNI3 p.R168Q mutation mediated up-regulation of FASN
To explore the effect of TNNI3 p.R168Q on FASN, we first conducted a co-IP assay and found that there was no interaction between cTnI and FASN [Supplementary Figure 7], which imply that this mutation might regulate the expression of FASN indirectly. Furthermore, we used bioinformatics methods to scan potential partners for cTnI. Consequently, an interaction between cTnI and EGFR was observed [Figure 5A], which suggested that the TNNI3 p.R168Q mutant might regulate FASN by affecting EGFR. To confirm the transactivation of EGFR, we detected the phosphorylation of AKT in vivo and in vitro. As expected, the expression of PI3K/AKT was increased in the Ad-Mut than in the Ad-WT group
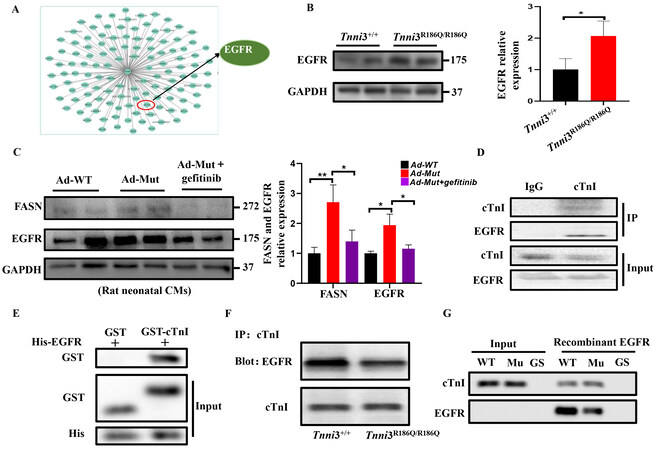
Figure 5. TNNI3 p.R168Q mutation regulates FASN expression through EGFR. (A) PPI network of TNNI3 generated using STRING 11.0. Nodes represent proteins. Edges represent PPIs. (B) EGFR protein levels measured by Western blot were detected in Tnni3+/+ and
DISCUSSION
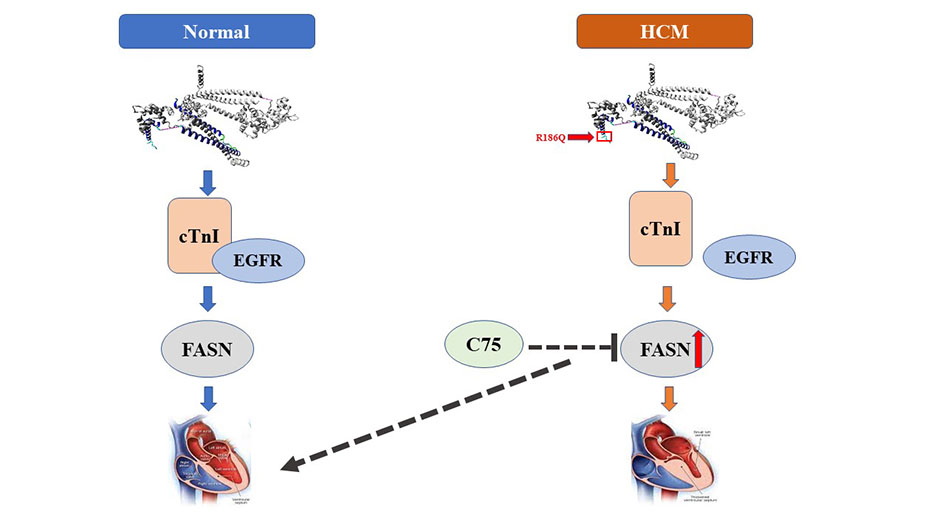
In the present study, we reported important novel findings demonstrating the role of the TNNI3 p.R186Q mutation and its detailed mechanism in HCM. First, using different approaches, we demonstrated that the TNNI3 p.R186Q mutation can induce the phenotype of cardiac hypertrophy in vivo and in vitro. In addition, KI mice bearing this mutation displayed aberrant fatty acid metabolism. Mechanistically, the TNNI3 p.R186Q mutation reduced the interaction between cTnI and EGFR, activated EGFR and increased the expression of FASN, which likely bring about aberrant fatty acid metabolism and the development of HCM. Finally, the FASN selective inhibitor C75 appeared to be effective in the management of TNNI3 p.R186Q-related HCM [Figure 6].
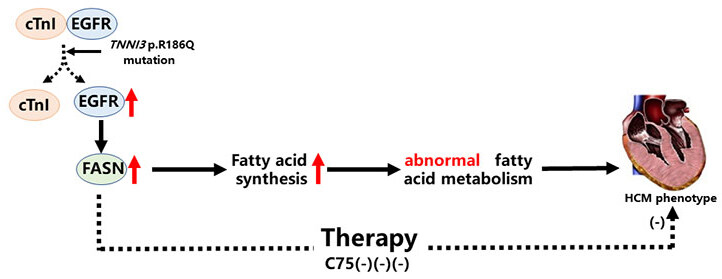
Figure 6. Schematic model of Tnni3R186Q/R186Q mouse related HCM.
In recent decades, several phenotypes of HCM have been caused by mutation-related landmarks, including thickening of the left ventricle or septal wall, disordered arrangement of myocardial cells, increased myocardial necrosis/fibrosis or inflammatory cell infiltration, and increased expression of hypertrophy markers (ANP, BNP, MYH6, MYH7)[16]. In this study, the KI mice at baseline showed thickening of the left ventricle and septal wall, obvious myocardial fibrosis, increased myocardial cell size, and increased expression of hypertrophy markers, which indicated that the mouse model was successfully established. It has been reported that most pathogenic mutations of the TNNI3 gene are located in the cTnI mobile domain region, which plays a key role in myocardial diastolic function. In view of the high-frequency occurrence of mutations in this region, some studies have suggested that there is a strict structure-function relationship in this domain[17], but others have indicated that these mutations are highly tolerated, thereby avoiding embryonic lethality and allowing these mutations to remain in the population[18]. The KI mice in this study survived normally to adulthood without embryonic lethality, further confirming the latter view.
FAs are the most important energy source in the heart[19], and abnormal FAs metabolism is critical to the pathogenesis of cardiac disease. Previous studies have demonstrated that some diseases including ischemic heart disease, heart failure, metabolic syndrome and type 2 diabetes all exhibit reduced oxidation of free FAs, which leads to the accumulation of FAs and corresponding changes in the structure of myocardial cells, such as myocardial fibrosis[20], decreased cardiac function, and pressure overload in cardiac hypertrophy[21-24]. In the present study, there was no change in the β-oxidation of FAs, but the synthesis of FAs was significantly increased in the myocardial tissue of Tnni3R186Q/R186Q mice, which also resulted in the accumulation of FAs in cardiomyocytes, and eventually triggered cardiac hypertrophy. Previous studies have found that mutations in sarcomere-related genes, as well as in metabolism-related energy-sensitive enzyme genes, can cause HCM[25,26]. Unexpectedly, this study found that TNNI3 sarcomere gene mutations can both directly cause HCM, and also cause HCM by further changing the metabolism of cardiomyocytes. Moreover, no change was observed in the TC, TG, LDL or HDL levels in the serum of KI mice, while the corresponding indicators of TC and TG in the myocardial tissue increased, further confirming the myocardial specificity of cTnI[27].
Previous studies have found that changes in protein-protein interactions between troponin complexes increase the severity of the disease[28]. Consistent with their findings, this study found that the TNNI3 p.R186Q mutation did not directly affect the structure of cTnI protein, but had a weakened effect on the interaction between cTnI and EGFR. EGFR plays an important role in regulating signaling pathways in cellular activities, such as proliferation, differentiation, and death[29]. Among them, FASN is one of the downstream targets of EGFR for regulating cell proliferation. FASN is a key enzyme involved in the de novo fatty acid synthesis pathway, which plays a very important role in cell FAs accumulation and affects many metabolic processes of cells. Many studies have shown that under pathological conditions, the overexpression of FASN is closely related to the development of cardiovascular disease, type 2 diabetes, many types of cancer and fibroproliferative actions[30-32]. Similarly, in this study, the overexpression of FASN directly caused the accumulation of FA in myocardial tissue. This is probably the main cause of HCM. Several groups have investigated that the activation of EGFR can directly increase the levels of intracellular FAs, and at the same time, if EGFR inhibitors are used, the content of intracellular free FAs can be significantly reduced[33,34]. Other studies also found that EGFR has a positive feedback regulatory effect on the expression of FASN. The activation of EGFR can promote an increase in FASN expression, and the increased expression of FASN can in turn promote the activation of EGFR, which strongly induces cell growth[35]. Likewise, in our KI hearts, the TNNI3 p.R186Q mutation may weaken the interaction between cTnI and EGFR, activate EGFR and further increase the expression of FASN, leading to FA accumulation.
Until now, there has been little evidence that drug treatment alone can fundamentally change the natural history of HCM or cause left ventricular remodeling[36]. For more than 30 years, no other HCM treatment drugs have been introduced[37]. In this study, we found that FASN inhibition of C75 can improve cardiomyocyte hypertrophy and fibrosis, which will help open more clinical options for HCM treatment in the future, and might represent a new treatment strategy for TNNI3 p.R186Q related HCM.
Several limitations should be considered in this study. First, most of the pathogenic variants identified in HCM patients are heterozygous mutations, but interestingly, the heterozygous mice in this study did not have an overt HCM phenotype. Therefore, although the mouse model provides insight into the mechanisms of human disease, inherent differences between human and mouse physiology must ultimately be considered when interpreting our findings. Second, we mainly focused on the effects of FASN inhibitors in the mouse model. Previous studies have shown that EGFR inhibitors can prevent myocardial hypertrophy and heart damage through a variety of pathways, and there is no specific inhibitor of the EGFR-FASN pathway thus far[38,39]. Therefore, EGFR inhibitors were not studied in this study and whether inhibition of EGFR could affect TNNI3 p.R186Q related HCM is still unknown. Further investigations will provide insight into these fascinating questions. The primary phenotype was analyzed in a very large number of mice, including 78 Rnni3R186Q/R186Q mice [Figure 1B]. Likewise, cardiac function was analyzed in 8 mice per genotype. However, some of the secondary phenotypes, such as myocardial histology and western blotting were analyzed only in 3 mice per genotype. Thus, the secondary data are considered provisional pending replication in additional studies.
In conclusion, this study reveals a novel mechanism in the development of TNNI3 p.R186Q related HCM by promoting abnormal fatty acid metabolism through FASN. Importantly, FASN inhibition is an interesting target for future research on TNNI3 p.R186Q related to HCM.
DECLARATIONS
Authors’ contributionsConceived, designed and coordinated the study: Hong K, Wan R
Conducted the experiments, analyzed the data and wrote the paper: Guo L, Su Y, Chen C
Contributed to manuscript revisions: Guo L, Su Y, Chen C, Zhou Q, Shen Y, Jiang Z, Yan X, Li X, Zhuo W, Peng X, Wan R, Hong K
All authors have read and approved the final version of the manuscript and agree to be responsible for its content.
Availability of data and materialsThe data supporting the results of this study can be obtained from the corresponding author upon reasonable request. For detailed methods, please refer to the online Supplementary Materials.
Financial support and sponsorshipThis work was supported by the National Natural Science Foundation of China (Grant No. 81960045 to Zhou Q, 82030018 to Hong K, 31971041 to Hong K) and Jiangxi Province Technology Innovation Guidance Program (20203AEI005, Hong K).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateAll animal care procedures and experiments were approved by the Animal Ethics and Experimentation Committee of Nanchang University and conformed to the “Guide for the Care and Use of Laboratory Animals” (revised 1996).
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
Supplementary MaterialsREFERENCES
1. Seidman CE, Seidman JG. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ Res 2011;108:743-50.
2. Ho CY, Day SM, Ashley EA, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the sarcomeric human cardiomyopathy registry (SHaRe). Circulation 2018;138:1387-98.
3. Ingles J, Goldstein J, Thaxton C, et al. Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ Genom Precis Med 2019;12:e002460.
4. Mogensen J, Murphy RT, Kubo T, et al. Frequency and clinical expression of cardiac troponin I mutations in 748 consecutive families with hypertrophic cardiomyopathy. J Am Coll Cardiol 2004;44:2315-25.
5. Morner S. Identification of the genotypes causing hypertrophic cardiomyopathy in northern Sweden. J Mol Cell Cardiol 2003;35:841-9.
6. Hu J, Xiang Li J, Hong K, Xin Hu J, Brugada P, Shu Cheng X. Hypertrophic cardiomyopathy and planned in vitro fertilization. Genetic testing and clinical evaluation. Herz 2012;37:447-52.
7. Carley AN, Taegtmeyer H, Lewandowski ED. Matrix revisited: mechanisms linking energy substrate metabolism to the function of the heart. Circ Res 2014;114:717-29.
8. Glenn DJ, Wang F, Nishimoto M, et al. A murine model of isolated cardiac steatosis leads to cardiomyopathy. Hypertension 2011;57:216-22.
9. Krishnan J, Suter M, Windak R, et al. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab 2009;9:512-24.
10. Menendez JA, Lupu R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin Ther Targets 2017;21:1001-16.
11. Che L, Pilo MG, Cigliano A, et al. Oncogene dependent requirement of fatty acid synthase in hepatocellular carcinoma. Cell Cycle 2017;16:499-507.
12. Reinecke H, Zhang M, Bartosek T, Murry CE. Survival, integration, and differentiation of cardiomyocyte grafts: a study in normal and injured rat hearts. Circulation 1999;100:193-202.
13. Zhou Q, Peng X, Liu X, et al. FAT10 attenuates hypoxia-induced cardiomyocyte apoptosis by stabilizing caveolin-3. J Mol Cell Cardiol 2018;116:115-24.
14. Peng X, Shao J, Shen Y, et al. FAT10 protects cardiac myocytes against apoptosis. J Mol Cell Cardiol 2013;59:1-10.
15. Thupari JN, Landree LE, Ronnett GV, Kuhajda FP. C75 increases peripheral energy utilization and fatty acid oxidation in diet-induced obesity. Proc Natl Acad Sci USA 2002;99:9498-502.
16. Hurtado-de-Mendoza D, Corona-Villalobos CP, Pozios I, et al. Diffuse interstitial fibrosis assessed by cardiac magnetic resonance is associated with dispersion of ventricular repolarization in patients with hypertrophic cardiomyopathy. J Arrhythm 2017;33:201-7.
17. Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, Potter JD. Mutations in troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function? J Mol Cell Cardiol 2010;48:882-92.
18. Ghashghaee N, Li KL, Solaro RJ, Dong WJ. Role of the C-terminus mobile domain of cardiac troponin I in the regulation of thin filament activation in skinned papillary muscle strips. Arch Biochem Biophys 2018;648:27-35.
19. Klip A, Sun Y, Chiu TT, Foley KP. Signal transduction meets vesicle traffic: the software and hardware of GLUT4 translocation. Am J Physiol Cell Physiol 2014;306:C879-86.
20. Legchenko E, Chouvarine P, Borchert P, et al. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci Transl Med 2018;10:eaao0303.
21. Saini-Chohan HK, Mitchell RW, Vaz FM, Zelinski T, Hatch GM. Delineating the role of alterations in lipid metabolism to the pathogenesis of inherited skeletal and cardiac muscle disorders: thematic review series: genetics of human lipid diseases. J Lipid Res 2012;53:4-27.
22. Wang ZV, Li DL, Hill JA. Heart failure and loss of metabolic control. J Cardiovasc Pharmacol 2014;63:302-13.
23. He L, Kim T, Long Q, et al. Carnitine palmitoyltransferase-1b deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity. Circulation 2012;126:1705-16.
24. Kienesberger PC, Pulinilkunnil T, Sung MM, et al. Myocardial ATGL overexpression decreases the reliance on fatty acid oxidation and protects against pressure overload-induced cardiac dysfunction. Mol Cell Biol 2012;32:740-50.
25. Chung H, Kim Y, Cho SM, et al. Differential contributions of sarcomere and mitochondria-related multigene variants to the endophenotype of hypertrophic cardiomyopathy. Mitochondrion 2020;53:48-56.
26. Baker ZN, Jett K, Boulet A, et al. The mitochondrial metallochaperone SCO1 maintains CTR1 at the plasma membrane to preserve copper homeostasis in the murine heart. Hum Mol Genet 2017;26:4617-28.
27. Ricchiuti V, Apple FS. RNA expression of cardiac troponin T isoforms in diseased human skeletal muscle. Clin Chem 1999;45:2129-35.
28. Nguyen S, Siu R, Dewey S, Cui Z, Gomes AV. Amino acid changes at arginine 204 of troponin I result in increased calcium sensitivity of force development. Front Physiol 2016;7:520.
29. Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017;9:52.
30. Cabarcas SM, Hurt EM, Farrar WL. Defining the molecular nexus of cancer, type 2 diabetes and cardiovascular disease. Curr Mol Med 2010;10:744-55.
31. Menendez JA, Vazquez-Martin A, Ortega FJ, Fernandez-Real JM. Fatty acid synthase: association with insulin resistance, type 2 diabetes, and cancer. Clin Chem 2009;55:425-38.
32. Jung MY, Kang JH, Hernandez DM, et al. Fatty acid synthase is required for profibrotic TGF-β signaling. FASEB J 2018;32:3803-15.
33. Zhang J, Song F, Zhao X, et al. EGFR modulates monounsaturated fatty acid synthesis through phosphorylation of SCD1 in lung cancer. Mol Cancer 2017;16:127.
34. Polonio-Alcalá E, Palomeras S, Torres-Oteros D, et al. Fatty acid synthase inhibitor G28 shows anticancer activity in EGFR tyrosine kinase inhibitor resistant lung adenocarcinoma models. Cancers 2020;12:1283.
35. Ali A, Levantini E, Teo JT, et al. Fatty acid synthase mediates EGFR palmitoylation in EGFR mutated non-small cell lung cancer. EMBO Mol Med 2018:10.
36. Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the american college of cardiology/american heart association joint committee on clinical practice guidelines. J Am Coll Cardiol 2020;76:e159-240.
37. Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with β-blockade or verapamil. Circ Heart Fail 2013;6:694-702.
38. Peng K, Tian X, Qian Y, et al. Novel EGFR inhibitors attenuate cardiac hypertrophy induced by angiotensin II. J Cell Mol Med 2016;20:482-94.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Guo L, Su Y, Chen C, Zhou Q, Shen Y, Jiang Z, Yan X, Li X, Zhuo W, Peng X, Wan R, Hong K. The TNNI3 p.R186Q mutation is responsible for hypertrophic cardiomyopathy via promoting FASN-stimulated abnormal fatty acid metabolism. J Cardiovasc Aging 2023;3:6. http://dx.doi.org/10.20517/jca.2022.29
AMA Style
Guo L, Su Y, Chen C, Zhou Q, Shen Y, Jiang Z, Yan X, Li X, Zhuo W, Peng X, Wan R, Hong K. The TNNI3 p.R186Q mutation is responsible for hypertrophic cardiomyopathy via promoting FASN-stimulated abnormal fatty acid metabolism. The Journal of Cardiovascular Aging. 2023; 3(1): 6. http://dx.doi.org/10.20517/jca.2022.29
Chicago/Turabian Style
Guo, Linjuan, Yuhao Su, Chen Chen, Qiongqiong Zhou, Yang Shen, Zhenhong Jiang, Xia Yan, Xiaoqing Li, Wen Zhuo, Xiaogang Peng, Rong Wan, Kui Hong. 2023. "The TNNI3 p.R186Q mutation is responsible for hypertrophic cardiomyopathy via promoting FASN-stimulated abnormal fatty acid metabolism" The Journal of Cardiovascular Aging. 3, no.1: 6. http://dx.doi.org/10.20517/jca.2022.29
ACS Style
Guo, L.; Su Y.; Chen C.; Zhou Q.; Shen Y.; Jiang Z.; Yan X.; Li X.; Zhuo W.; Peng X.; Wan R.; Hong K. The TNNI3 p.R186Q mutation is responsible for hypertrophic cardiomyopathy via promoting FASN-stimulated abnormal fatty acid metabolism. J. Cardiovasc. Aging. 2023, 3, 6. http://dx.doi.org/10.20517/jca.2022.29
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 15 clicks
Cite This Article 15 clicks

 Like This Article 10
likes
Like This Article 10
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.