
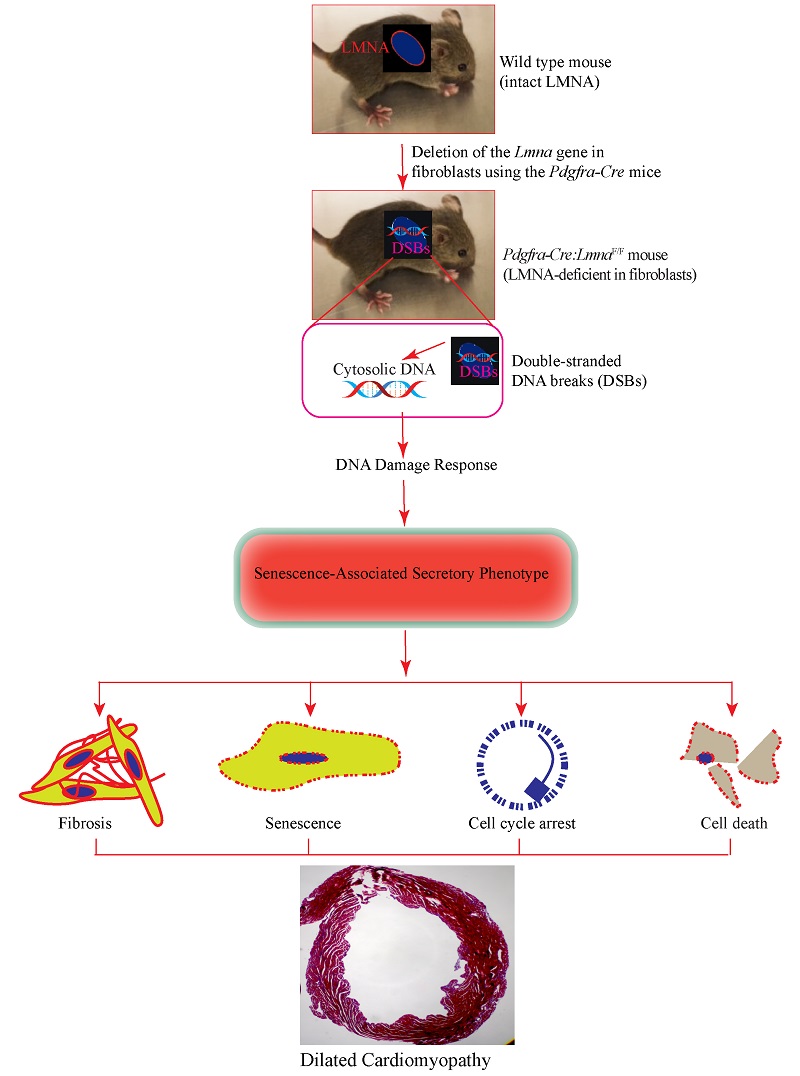
Deletion of the Lmna gene in fibroblasts causes senescence-associated dilated cardiomyopathy by activating the double-stranded DNA damage response and induction of senescence-associated secretory phenotype

 , ...
, ... Abstract
Introduction: Mutations in the LMNA gene, encoding Lamin A/C (LMNA), are established causes of dilated cardiomyopathy (DCM). The phenotype is typically characterized by progressive cardiac conduction defects, arrhythmias, heart failure, and premature death. DCM is primarily considered a disease of cardiac myocytes. However, LMNA is also expressed in other cardiac cell types, including fibroblasts.
Aim: The purpose of the study was to determine the contribution of the fibroblasts to DCM caused by LMNA deficiency.
Methods and Results: The Lmna gene was deleted by crossing the platelet-derived growth factor receptor α-Cre recombinase (Pdgfra-Cre) and floxed Lmna (LmnaF/F) mice. The LMNA protein was nearly absent in ~80% of the cardiac fibroblasts and ~25% of cardiac myocytes in the Pdgfra-Cre:LmnaF/F mice. The Pdgfra-Cre:LmnaF/F mice showed an early phenotype characterized by cardiac conduction defects, arrhythmias, cardiac dysfunction, myocardial fibrosis, apoptosis, and premature death within the first six weeks of life. The Pdgfra-Cre:LmnaWild type/F
Conclusion: Deletion of the Lmna gene in fibroblasts partially recapitulates the phenotype of the LMNA-associated DCM, likely through induction of double-stranded DNA breaks, activation of the DDR pathway, and induction of expression of the SASP proteins. The findings indicate that the phenotype in the LMNA-associated DCM is the aggregate consequence of the LMNA deficiency in multiple cardiac cells, including cardiac fibroblasts.
One sentence summary: Cardiac fibroblasts contribute to the pathogenesis of DCM - associated with LMNA deficiency through activation of the senescence-associated secretory phenotype.
Keywords
INTRODUCTION
Heart failure is a major cause of mortality and morbidity in the world, affecting over 60 million people globally[1,2]. Hereditary cardiomyopathies are major causes of heart failure and sudden cardiac death[1,3]. Hereditary dilated cardiomyopathy (DCM) comprises a genetically heterogeneous group of disorders characterized by cardiac dilatation and systolic dysfunction[4]. Mutations in the LMNA gene, encoding nuclear envelope protein Lamin A/C (LMNA), are important causes of hereditary DCM[4-7].
LMNA is expressed ubiquitously in almost all differentiated cells, including cardiac myocytes and fibroblasts[8]. Mutations in the LMNA gene cause a diverse array of clinical phenotypes, in addition to DCM, which are collectively referred to as laminopathies[9,10]. In accord with the ubiquitous expression of the LMNA gene in various cell types, the phenotypes in laminopathies are the aggregate consequences of the expression of the causal mutations in multiple cell types, tissues, and organs.
Cardiac involvement in laminopathies is common and typically manifests with DCM, atrial and ventricular conduction defects, supraventricular and ventricular arrhythmias, and sudden cardiac death[6,7]. DCM is the major cause of morbidity and mortality in laminopathies, particularly in the subset that predominantly involves the striated muscles[5-7,11-13]. LMNA mutations have been identified in up to 10% of the patients with familial DCM[6,7]. There is no specific therapy for laminopathies, including DCM- associated with the LMNA mutations.
The heart is a cellularly heterogeneous organ. Cardiac myocytes are considered the main cell type involved in the LMNA-associated DCM, a notion that is in accord with cardiomyopathies being considered mainly the diseases of the cardiac myocytes. Consequently, the contributions of other cardiac cell types, such as cardiac fibroblasts that also express the mutant LMNA to the pathogenesis of DCM, have remained unexplored. The purpose of the present study was to delineate the contribution of cardiac fibroblasts to the pathogenesis of DCM observed in laminopathies. Therefore, the Lmna gene was deleted in fibroblasts in mice upon expression of the Cre recombinase under the transcriptional controls of the platelet-derived growth factor receptor-α (Pdgfra) locus, which tags most of the fibroblasts in the heart. The ensuing cardiac phenotype was characterized in the Lmna heterozygous and homozygous mice.
METHODS
Data sharing and availability: RNA-Seq data have been deposited in the public database GEO (Available online: https://www.ncbi.nlm.nih.gov/gds, GSE199078). Additional data about the present article are available from the corresponding author upon request.
Regulatory approvals: Animal procedures were in full compliance with the NIH Guidelines for the Care and Use of Laboratory Animals and approved by the Animal Care and Use Committee of the University of Texas Health Science Center - Houston (AWC-21-0015).
Anesthesia and euthanasia: Anesthesia was induced upon inhalation of 3% isoflurane and was maintained by inhalation of 0.5%-1% isoflurane throughout the procedures. Mice were euthanized with CO2 gas inhalation followed by cervical dislocation.
Deletion of the Lmna gene in fibroblasts: To delete the Lmna gene in fibroblasts, mice carrying floxed
Survival: Kaplan-Meier survival plots for the WT, Pdgfrα-Cre, Pdgfrα-Cre:LmnaW/F, and Pdgfrα-Cre:LmnaF/F mice were constructed, and the survival rates were compared by the log-rank test.
Bodyweight: Body weight was measured weekly after two weeks of age in each mouse and compared among the WT, Pdgfrα-Cre, Pdgfrα-Cre:LmnaW/F, and Pdgfrα-Cre:LmnaF/F mice.
Metabolic panel: Blood was collected from the retroorbital vein for the assessment of plasma metabolic panels, comprised of indices of kidney and liver functions as well as electrolytes, glucose, and lipid levels, in mice kept fast overnight.
Echocardiography: Cardiac size and function were assessed in age- and sex-matched mice in each of the four experimental groups by 2D, M mode, and Doppler echocardiography, as published using a Vevo 1100 ultrasound imaging system equipped with a 22-55 MHz MicroScan transducer (MS550D) (FUJIFILM VisualSonics Inc., Toronto, ON, Canada)[17-19].
Echocardiography was performed in six weeks old WT, Pdgfrα-Cre, Pdgfrα-Cre:LmnaW/F, and
In brief, echocardiography was performed in mice lightly anesthetized with 0.5%-1% isoflurane inhalation. B-modes-guided M modes parasternal short-axis images were obtained in the supine position at the level of the tip of the papillary muscles. Left ventricular anterior wall thickness (LVAWT), LV posterior wall thickness (LVPWT), LV end-diastolic diameter (LVEDD), and LV end-systolic diameter (LVESD) were measured in 5 to 6 cardiac cycles using the leading-edge method, and the mean value of the measurements was calculated. LV fractional shortening and LV mass were calculated from the measured indices[20]. Echocardiographic measurements were indexed to body weight, as body weight is a strong determinant of indices of cardiac size, such as LVEDD, LVESD, and LVM. The corrected indices were defined as LVEDDI, LVESDI, and LVMI. Image acquisition and data analysis were performed without knowledge of the mouse genotype.
Electrocardiography (ECG): Two-lead precordial surface ECG was recorded by inserting subcutaneous needle electrodes into the upper right and left precordial area. During the recording, mice were kept lightly anesthetized using 0.5%-1% isoflurane and under constant monitoring on a 37 °C heated pad. Leads were connected to a PowerLab 4/30 System using an Animal Bio Amp module (ADInstruments, Colorado Springs, CO, USA). The rhythm was recorded and visualized using LabChart7 Software without knowledge of the genotype.
Myocyte cross-sectional area (CSA): To determine cardiac myocyte CSA, thin myocardial sections were stained with wheat germ agglutinin (WGA) to mark the extracellular matrix and myocyte boundaries, as published[17,18]. The total area unstained with WGA was determined using the Image J software (Available online: https://imagej.net) and used to calculate the myocyte CSA. Approximately 20,000 cells per mouse and 4 to 5 mice per genotype were analyzed.
Myocardial Fibrosis: Myocardial fibrosis was detected upon staining of thin myocardial sections with picrosirius red, as published[17,18]. Collagen volume fraction (CVF) was calculated as the percent area of the myocardial section stained positive for picrosirius red using the Image J software in 10 high magnification microscopic fields (×40) per section, in 6 sections per heart, and a minimum of 4 mice per each experimental group, as published[17,18].
Myocardial adipocytes: To detect adipocytes in the heart, thin myocardial sections were prepared from the paraffin-embedded tissues and subjected to antigen retrieval upon treatment with 10 mM of boiling sodium citrate (pH 6.0). The sections were incubated overnight with an anti-perilipin 1 (PLIN1) antibody to identify lipid-containing adipocytes (Cell Signaling Technology, cat. # 9349), as published[19]. Further, sections were washed with PBS, incubated with the secondary antibody conjugated to Alexa 594, and counterstained with 4′,6 Diamidino-2-phenylindole dihydrochloride (DAPI, Sigma-Aldrich St Louis, MO; cat. # D8417) at
TUNEL assay: Cells undergoing apoptosis in the myocardium were detected using the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay and the In-Situ Cell Death Detection Fluorescein Kit, as published[18,21]. The number of cells stained positive for the TUNEL assay was determined in approximately, 10,000 cells per heart and in 5 to 6 hearts per genotype, and the percentages were compared.
Isolation of cardiac fibroblasts: Mice were anesthetized with an intraperitoneal injection of pentobarbital (62 mg/kg, ip) and anti-coagulated upon IP administration of 200 units of heparin. The heart was harvested, rinsed in a cold PBS buffer, and cannulated through the ascending aorta. The cannula was positioned above the aortic valves and connected to a retrograde perfusion system. The heart was perfused with PBS at a constant rate of 4 mL/min until blood was fully rinsed. Then, collagenase II (Worthington, Lakewood, NJ) was added to the perfusion buffer at 25 mg/25 mL of 250 units/mL solution, and the heart was perfused until complete softening of the myocardium (typically for about 8 minutes). The heart was then minced into small pieces in the collagenase buffer and cells were dissociated by gentle pipetting. Upon complete dissociation of the tissue, the collagenase activity was stopped by adding calf serum (10% in the final volume), and the cell suspension was passed through a 40 µm mesh cell strainer to remove the debris. The cells were then centrifuged at 300 g for 10 min at 4 °C, and the pellet was resuspended in a MACS buffer (Miltenyi Biotec, Bergisch Gladbach, Germany; cat. # 130-091-221) and washed with PBS. The cells were then incubated with an antibody against PDGFRA (cat. # 17-1401-81, eBioscience) and a cocktail of mouse lineage antibodies (cat. # 561301, BD Pharmingen, BD Horizon™ V450 Mouse Lineage Antibody Cocktail, with Isotype Control) for 1 h at 4 °C in the dark. Following antibody treatment, 4′,6 Diamidino-2-phenylindole dihydrochloride (DAPI, cat. # D8417, Sigma-Aldrich) was added at 0.1 mg/mL to the cell suspension for 10 minutes. The unbound antibodies and excess DAPI were removed by washing the cells twice in the MACS buffer. The cell suspension was passed through a mesh strainer before sorting through a FACS-Aria flow cytometer (BD Pharmingen, San Diego, CA). Cells were sorted to isolate living (DAPIneg) and hematopoietic lineage negative (Linneg) cells that expressed PDGFRA (PDGFRApos cells). Unlabeled cells, as well as cells stained with the appropriate isotype IgG controls (cat. # 17-4321, eBioscience, APC Rat IgG2a K Isotype Control), were included as controls.
Cardiac myocyte isolation: Cardiac myocytes were isolated as published[17-19,22]. In brief, the heart was excised from the anesthetized mice and perfused retrogradely with the digestion collagenase buffer at a concentration of 2.4 mg/mL concentration at a flow rate of 4 mL/min (Worthington cat. # LS004176). Following the enzymatic digestion, the heart was minced in a 10% calf serum, 12.5 μM CaCl2, and 2 mM ATP buffer, and the cell suspension was filtered through a 100 µm cell strainer. Cardiac myocytes per precipitated by centrifugation at 20 g and re-introduction of an increasing concentration of calcium in a stepwise fashion with a final concentration of 900 μM added to the stop buffer. Then, the cardiac myocytes were suspended in a Qiazol reagent (Qiagen cat. # 79306) for RNA extraction, in a protein extraction buffer for immunoblotting, or fresh frozen in isopentane for immunofluorescence studies.
Immunoblotting. Total protein was extracted from cardiac fibroblasts, quantified by the Bradford assay, and was used for immunoblotting, as published[18,21,23]. Approximately 50 µg aliquots of the protein extracts were loaded onto polyacrylamide gels, separated by electrophoresis, transferred to a nitrocellulose membrane, and probed with the desired antibody. The primary and secondary antibodies used are listed in Supplementary Table 1.
Immunofluorescence: To detect the expression and localization of the protein of interest, thin myocardial frozen sections were prepared and probed with specific primary antibodies as published[19]. The antibodies used for the immunofluorescence studies are listed in Supplementary Table 1.
Senescence β-galactosidase staining: Senescence β-galactosidase staining kit (Cell Signaling Technology, cat. # 9860) was used to detect β-galactosidase activity according to the manufacturer’s protocol. Briefly, thin fresh frozen myocardial sections were fixed using the 1X fixative solution for 15 min and washed twice with 1X PBS. The fixed sections were incubated with the β-galactosidase solution in a sealed humid box overnight at 37 °C in a dry incubator (no CO2). The sections were rinsed with PBS, counterstained with Eosin, and mounted using 70% glycerol for long-term storage. The images were taken using a 20× magnification using an Olympus BX40 microscope.
RT-PCR: To detect and compare transcript levels of the selected genes, RT-PCR was performed as published[17,19]. In brief, total RNA was extracted using the miRNeasy Mini Kit (cat. # 217004, Qaigen Inc.), treated with DNase, and reverse transcribed into cDNA using the High-Capacity cDNA Reverse Transcription Kit (Life Tech, cat. # 4368814). The RT-PCR was performed using the TaqMan gene expression assays or SYBR Green specific primers. Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) mRNA levels were used as loading controls for normalization. Gene expression was compared using the
Sequencing (RNA-Seq): RNA-Seq was performed on ribosome-depleted RNA isolated from cardiac fibroblasts, as published, with some modifications[18,21,23]. In brief, cardiac fibroblasts were isolated from the ventricular tissues and used for total RNA extraction. The RNA samples that had an RNA Integrity Number (RIN) of > 8 were used to generate strand-specific RNA sequencing libraries. The sequencing reaction was performed on an Illumina instrument, and 150 bp paired-end reads were generated and aligned to the mouse reference genome build mm10 using HISAT2[24]. The GENCODE program (Available online: https://www.gencodegenes.org/mouse/) was used to annotate the aligned read pairs. Read counts were determined using the featureCounts upon RUV normalization, and the differentially expressed genes (DEGs) were identified using the DESeq2 program in the R package (Available online: https://www.r-project.org/)[25,26]. A Q-value (Benjamini-Hochberg FDR-adjusted P-value) of < 0.05 was used to define the DEGs[27].
Pathway analysis: The RNA-seq data were analyzed using GSEA to identify gene sets that were enriched in the RNA-Seq data, as compared to the curated gene sets for Hallmark and the canonical pathways of the Molecular signature database (MSigDB). An FDR of < 0.05 was used to identify significantly enriched pathways.
To identify transcriptional regulators (TRs) that were predicted to be activated or suppressed, the DEGs were overlapped with the curated genes in each transcriptional pathway using the Ingenuity pathway analysis (IPA) software. The predicated TRs were defined based on a P-value of < 0.05 and a Z score of greater than +2 or less than -2.
Statistical analysis: Data were analyzed for a Gaussian distribution using the Shapiro-Wilk normality test. Data following the Gaussian distribution were compared by the t-test or one-way ANOVA followed by Bonferroni pairwise comparisons. The data that deviated from a Gaussian distribution and the categorical data were analyzed by the Kruskal-Wallis and the Chi-Square test, respectively. Survival rates were analyzed by the Log-rank test. Statistical analyses were performed using Graph pad Prism 8 or STAT IC, 15.1.
RESULTS
The efficiency of deletion of the Lmna gene in cardiac fibroblasts: To determine the efficiency of deletion of the Lmna gene, cardiac fibroblasts were isolated from the WT, Pdgfra-Cre, Pdgfra-Cre:LmnaW/F, and Pdgfra-Cre:LmnaF/F mouse hearts and the protein extracts from these cells were probed with an anti-LMNA antibody. LMNA protein levels were reduced by about 89.21 ± 5.62 in the cardiac fibroblast isolated from the Pdgfra-Cre:LmnaF/F and 17.12 ± 15.23 in the Pdgfra-Cre:LmnaW/F mice as compared to the WT or

Figure 1. Fidelity of conditional deletion of the Lmna gene in cardiac fibroblasts. (A) Immunoblots showing LMNA protein levels in cardiac fibroblasts isolated from the wild type (WT), Pdgfra-Cre, Pdgfra-Cre:LmnaW/F, and Pdgfra-Cre:LmnaF/F mice. The upper panel shows the LMNA protein. Blots representing cardiac troponin T (TNNT2) and vimentin (VIM), as markers of cardiac myocytes and fibroblasts, are included to assess the fidelity of the cell isolation. Blots representing GAPDH are included as loading controls. (B) Quantitative data on the LMNA protein levels corresponding to the blots in panel A. (C) Immunofluorescence staining of isolated cardiac fibroblasts for the expression of PDGFRA (upper panels) and LMNA (middle panel) and the overlay (lower panel). (D) Panel D the percentages of the isolated non-myocyte cells expressing PDGFRA. (E) Percentage of the PDGFRA expressing cells in which the LMNA protein was absent. In all analyses in the present and subsequent Figures, pairwise P-values were presented only when the P-value by the one-way ANOVA test was significant.
To complement the findings, isolated cardiac fibroblasts were also co-immunostained with antibodies against LMNA and PDGFRA, as well as DAPI, the latter to stain the nuclei. Approximately 73.1% ± 7.1% to 79.6% ± 3.7% of the isolated non-myocyte cardiac cells expressed PDGFRA showing no difference among the genotypes, as detected by immunofluorescence staining [Figure 1C and D]. All isolated non-myocyte cardiac cells expressing PDGFRA also expressed LMNA in the WT and Pdgfra-Cre groups, consistent with the ubiquitous expression of the LMNA in all post-natal cells [Figure 1C and E]. Expression of the LMNA was absent in 8.2% ± 4.8% of the non-myocyte cells expressing PDGFRA in the Pdgfra-Cre:LmnaW/F mice, which is reflective of one copy of the Lmna gene being intact and expressing the LMNA protein. The LMNA expression was not detected in 79.5% ± 5.8% of the non-myocyte cells that stained positive for the PDGFRA in the Pdgfra-Cre:LmnaF/F mice, indicating effective ablation of both copies of the Lmna gene in the isolated cardiac fibroblasts in the latter genotype [Figure 1C and E].
The Pdgfra locus is transcriptionally active in a small subset of cardiac myocytes during embryonic development but not in the adult heart[16]. To determine whether deletion of the Lmna gene under the transcriptional control of the Pdgfra locus (Pdgfra-Cre mice) also affects the expression of the LMNA protein in cardiac myocytes, protein extracts from adult mouse cardiac myocytes were probed for the expression of the LMNA protein by immunoblotting and immunofluorescence staining. The LMNA protein levels were unchanged in the Pdgfra-Cre:LmnaW/F mouse myocytes but were reduced by ~30% in the Pdgfra-Cre:LmnaF/F mouse myocytes [Figure 2A and B]. To assess the purity of the isolates, the membranes were probed with antibodies against TNNT2 and VIM, representing cardiac myocytes and fibroblasts, respectively. As would be expected, TNNT2 but not VIM was expressed in the cardiac myocyte protein isolates, as detected by immunoblotting [Figure 2A]. Immunofluorescence staining of isolated cardiac myocytes identified the absence of the LMNA protein in 29.28% ± 1.18% of cardiac myocytes

Figure 2. Fortuitous deletion of the Lmna gene in a subset of cardiac myocytes. (A) Immunoblots showing LMNA protein levels in cardiac myocytes isolated the WT, Pdgfra-Cre, Pdgfra-Cre:LmnaW/F, and Pdgfra-Cre:LmnaF/F mice. As in Figure 1, blots representing TNNT2 and VIM are included to assess the fidelity of the cell isolation. Blots representing GAPDH are included as controls for the loading conditions. (B) Quantitative data corresponding to the blots shown in F, showing about 30% reduction in the LMNA protein levels in the cardiac myocytes isolated from the Pdgfra-Cre:LmnaF/F mice, whereas the LMNA levels were unchanged in the myocytes isolated from the Pdgfra-Cre and Pdgfra-Cre:LmnaW/F mice. (C) Immunofluorescence staining of isolated cardiac myocytes for the PCM1 expression (upper panel), which tags cardiac myocyte nuclei in the heart, LMNA (middle panel), and the overlay (lower panel). (D) Quantitative data showing the percentages of cardiac myocytes, identified by the expression of PCM1, also express the LMNA proteins.
Growth and survival: The Pdgfra-Cre:LmnaF/F mice exhibited growth retardation as evidenced by smaller body weight as compared to the WT and Pdgfra-Cre mice, whereas the Pdgfra-Cre:LmnaW/F mice had a normal growth rate [Figure 3A and B]. Likewise, the Pdgfra-Cre:LmnaF/F mice exhibited increased mortality starting at around 3-4 weeks of age and reaching 100% mortality by two months of age [Figure 3C]. The Pdgfra-Cre:LmnaW/F mice also showed increased mortality, albeit later in life and reaching mortality of ~50% at 18 months of age [Figure 3D], whereas the WT and Pdgfra-Cre mice had normal survival rates.

Figure 3. Gross phenotype and survival. (A) Gross morphology of the WT, Pdgfra-Cre, Pdgfra-Cre:LmnaW/F, and Pdgfra-Cre:LmnaF/F mice at six weeks of age. (B) Growth curves of the mice in the experimental groups up to six weeks of age. (C) Kaplan-Meier survival plots show the survival rates up to ~2 months of age, the longest survival time of the Pdgfra-Cre:LmnaF/F mice. (D) Kaplan-Meier survival plots show the survival rates up to 20 months of age. Log-rank test p-value is shown. (E-G) Heart weight (E), body weight (F), and heart weight to body weight ratio (HW/BW) of the WT, Pdgfra-Cre, Pdgfra-Cre:LmnaW/F, and Pdgfra-Cre:LmnaF/F mice at six weeks of age. (H) Wheat Germ Agglutinin (WGA) stained thin myocardial sections used to calculate myocyte cross-sectional area (CSA). I. Graph representing myocyte CSA indexed to heart weight (CSAI) in the control and experimental groups.
Metabolic panel: Plasma levels of electrolytes, glucose, and indices of kidney and liver function were comparable among the WT, Pdgfra-Cre:LmnaW/F, and Pdgfra-Cre:LmnaF/F mice, except for plasma levels of glucose, which was reduced in the Pdgfra-Cre:LmnaF/F mice [Supplementary Table 2].
Cardiac size and function: The heart weight was smaller in the Pdgfra-Cre:LmnaF/F mice, reflective of the smaller body size, while the heart weight/body weight ratio was increased in the Pdgfra-Cre:LmnaF/F mice
LV size and function were assessed by echocardiography at six weeks of age in all four groups and
Echocardiographic phenotype at six weeks of age
| WT | Pdgfra-Cre | Pdgfra-Cre:LmnaW/F | Pdgfra-Cre:LmnaF/F | P (ANOVA) | |
| N | 11 | 10 | 10 | 12 | NA |
| M/F | 6/5 | 5/5 | 5/5 | 6/6 | 0.995^ |
| Age (days) | 41.27 ± 1.60 | 42.3 ± 2.10 | 43.50 ± 4.20 | 39.01 ± 1.03 | 0.058 |
| Body weight (g) | 21.59 ± 3.40 | 22.58 ± 3.00 | 21.83 ± 2.80 | 7.64 ± 1.80#*& | < 0.0001 |
| HR (bpm) | 468 ± 28 | 477 ± 50 | 516 ± 68 | 468 ± 53 | 0.292 |
| AWT (mm) | 0.57 ± 0.07 | 0.64 ± 0.09 | 0.55 ± 0.04 | 0.43 ± 0.06#*& | < 0.0001 |
| LVPWT (mm) | 0. 58 ± 0.69 | 0. 61 ± 0.11 | 0.55 ± 0.05 | 0.41 ± 0.06#*& | < 0.0001 |
| LVEDD (mm) | 3.68 ± 0.17 | 3.62 ± 0.30 | 3.49 ± 0.36 | 2.97 ± 0.28#*& | < 0.0001 |
| LVEDDI (mm/g) | 0.17 ± 0.02 | 0.16 ± 0.02 | 0.16 ± 0.01 | 0.40 ± 0.08#*& | < 0.0001 |
| LVESD (mm) | 2.20 ± 0.16 | 2.16 ± 0.28 | 2.21 ± 0.35 | 2.12 ± 0.35 | 0.915 |
| LVESDI (mm/mg) | 0.10 ± 0.01 | 0.10 ± 0.01 | 0.10 ± 0.01 | 0.29 ± 0.07#*& | < 0.0001 |
| FS (%) | 40.20 ± 3.49 | 40.39 ± 3.71 | 37.07 ± 4.27 | 28.82 ± 6.59#*& | < 0.0001 |
| LV Mass (mg) | 59.28 ± 13.09 | 55.16 ± 10.89 | 47.42 ± 11.65 | 25.14 ± 5.46#*& | < 0.0001 |
| LVMI (mg/g) | 2.54 ± 0.32 | 2.64 ± 0.63 | 2.17 ± 0.46 | 3.34 ± 0.48#*& | < 0.0001 |
Echocardiographic indices of cardiac size and function in 15-month-old Pdgfra-Cre:LmnaW/F mice
| WT | Pdgfra-Cre | Pdgfra-Cre:LmnaW/F | P (ANOVA) | |
| N | 11 | 11 | 14 | |
| M/F | 4/7 | 6/4 | 7/7 | 0.670^ |
| Age (month) | 14.33 ± 2.01 | 14.50 ± 0.81 | 15.56 ± 2.16 | 0.125 |
| Body weight (g) | 40.16 ± 4.97 | 39.151 ± 5.52 | 35.77 ± 4.21 | 0.329 |
| HR (bpm) | 468 ± 45 | 459 ± 37 | 421 ± 25 | 0.582 |
| AWT(mm) | 0.68 ± 0.08 | 0.63 ± 0.03 | 0.67 ± 0.10 | 0.356 |
| LVPWT(mm) | 0. 75 ± 0.08 | 0. 64 ± 0.07 | 0.70 ± 0.07 | 0.080 |
| LVEDD (mm) | 4.08 ± 0.38 | 4.15 ± 0.44 | 4.25 ± 0.38 | 0.534 |
| LVEDDI(mm/g) | 0.10 ± 0.01 | 0.11 ± 0.01 | 0.12 ± 0.01#* | 0.0003 |
| LVESD (mm) | 2.46 ± 0.40 | 2.58 ± 0.37 | 2.89 ± 0.41#* | 0.036 |
| LVESDI (mm) | 0.07 ± 0.01 | 0.06 ± 0.01 | 0.08 ± 0.01#* | < 0.0001 |
| FS (%) | 39.30 ± 5.67 | 38.00 ± 3.90 | 32.20 ± 4.91#* | 0.002 |
| LV Mass (mg) | 84.00 ± 13.56 | 77.02 ± 19.60 | 88.70 ± 26.01 | 0.460 |
| LVMI (mg/g) | 2.09 ± 0.23 | 1.94 ± 0.40 | 2.46 ± 0.50* | 0.020 |
Cardiac arrhythmias: Cardiac rhythm, monitored for about an hour in each mouse (8-12 mice per group), was notable for the presence of supraventricular and ventricular tachycardia as well as atrioventricular (AV) blocks in the Lmna-deficient mice [Figure 4]. Specifically, 4/12 Pdgfra-Cre:LmnaF/F mice, monitored at six weeks of age, developed atrial fibrillation, 3/12 had non-sustained ventricular tachycardia and 1/12 had a narrow QRS tachycardia with apparent retrograde p wave [Figure 4]. The Pdgfra-Cre:LmnaW/F mice did not show notable arrhythmias at six weeks of age; however, they exhibited ventricular arrhythmias and AV blocks, including complete heart block at 12-18 months of age [Figure 4].

Figure 4. Electrocardiographic recordings. Two-channel cardiac rhythm recordings are shown in the WT, Pdgfra-Cre, Pdgfra-Cre:LmnaW/F, and Pdgfra-Cre:LmnaF/F mice. The Pdgfra-Cre:LmnaW/F mice showed second and third-degree atrioventricular blocks after one year of age (third set of panels). The Pdgfra-Cre:LmnaF/F mice exhibited runs of wide complex and narrow complex tachycardia episodes at 6 weeks of age (the lower panels).
Myocardial fibrosis: CVF, calculated from picrosirius red-stained thin myocardial sections, comprised

Figure 5. Myocardial fibrosis. (A) Representative images of Low (upper) and high (lower) magnification of thin myocardial sections stained for picrosirius red, which illustrates increased myocardial fibrosis in the Pdgfra-Cre:LmnaF/F mice at six weeks of age. (B) Quantitative data showing collagen volume fraction (CVF) in the experimental and control groups. (C) Immunoblot showing levels of latent and mature TGFβ1 in the control and experimental groups, which were markedly increased in the Pdgfra-Cre:LmnaW/F and Pdgfra-Cre:LmnaF/F mice. (D) Quantitative data representing the blot in panel C. (E) Transcript levels of selected markers of myocardial fibrosis, quantified by RT-PCR, in the control and experimental groups. (F) Thin myocardial sections stained for picrosirius red in the Pdgfra-Cre:LmnaW/F and control mice, showing increased myocardial fibrosis. (G) Quantitative data showing increased CVF in the Pdgfra-Cre:LmnaW/F mouse myocardium as compared to the WT and Pdgfra-Cre mice at one year of age. (H) Transcript levels of selected markers of fibrosis, showing increased levels of selected markers but not Tgfb1 in the myocardium of one-year-old Pdgfra-Cre:LmnaW/F mice.
CVF was also quantified in 12-18 months old Pdgfra-Cre:LmnaW/F mice, which comprised 5.3% ± 3.0% of the myocardium, as opposed to 1.7 ± 0.2 and 1.9% ± 0.1% in the WT and Pdgfra-Cre mice, respectively [Figure 5D and E]. Similarly, transcript levels of Col1a1, Col3a1, Postn, and Ctgf (Ccn2) were increased in the Pdgfra-Cre:LmnaW/F mouse hearts, while levels of Col5a2, Pcolce, Tgfb1, and Tgfb2 were unchanged [Figure 5H].
Myocardial apoptosis: The number of myocardial cells undergoing apoptosis, detected by the TUNEL assay, was increased in the myocardium of Pdgfra-Cre:LmnaF/F mice at six weeks of age, as compared to the WT, Pdgfra-Cre, and Pdgfra-Cre:LmnaW/F mice [Figure 6A and B]. Likewise, transcript levels of several genes involved in apoptosis, namely Bnip3, Gadd45b, Gadd45g, and Bcl2l11, were increased in the myocardium of Pdgfra-Cre:LmnaF/F mice, whereas those of Bcl2, Bbc3, and Bax were unchanged [Figure 6C]. Analysis of the Pdgfra-Cre:LmnaW/F mice at an older age also showed an increased number of the myocardial cells that stained for the TUNEL assay as well as increased transcript levels of Bax, Bbc3, Bcl2, Gadd45g, and Bcl2l11 but not Bnip3 and Gadd45b [Figure 6D-F].

Figure 6. Myocardial apoptosis. (A) The three panels show thin myocardial sections stained for the TUNEL assay. The inserts show a high magnification view of the selected areas marked with a red box. The data are from six weeks old mice. (B) Quantitative data showing the percentages of the TUNEL positive cells in the experimental and control groups at six weeks of age. (C) Transcript levels of selected markers of apoptosis, quantified by RT-PCR, in the control and the experimental groups. (D) Thin myocardial sections were stained for the TUNEL assay in the one-year-old Pdgfra-Cre:LmnaW/F and control mice. (E) Quantitative data showing an increased number of nuclei stained positive for the TUNEL assay in the one-year Pdgfra-Cre:LmnaW/F mice, as compared to the WT and the Pdgfra-Cre mice. (F) Transcript levels of selected markers of apoptosis, showing increased levels of the selected genes in the one-year-old Pdgfra-Cre:LmnaW/F mice, as compared to the control groups.
Myocardial adipocytes: To detect adipocytes in the myocardium, thin myocardial sections were stained with an antibody against perilipin 1 (PLIN1), which marks the triglyceride-laden fat cells. The number of adipocytes was increased in the Pdgfra-Cre:LmnaF/F mouse hearts as compared to the WT, Pdgfra-Cre, and Pdgfra-Cre:LmnaW/F mouse hearts [Figure 7A and B]. In addition, transcript levels of Pparg, Cebpa, and Dgat1, major regulators of adipogenesis, were increased in the Pdgfra-Cre:LmnaF/F mouse hearts, whereas levels of Dgat2, and Adipoq transcripts were unchanged [Figure 7C]. The heterozygous Lmna mice were also analyzed for adipogenesis at age 12-18 months of age, which also showed an increased number of myocardial cells expressing PLIN1 as compared to the WT or Pdgfra-Cre mice [Figure 7D and E]. Likewise, levels of the Cebpa, a major regulator of adipogenesis, were markedly increased in the Pdgfra-Cre:LmnaW/F mouse hearts [Figure 7F].

Figure 7. Myocardial adipocytes. (A) Thin myocardial sections were stained for the expression of perilipin 1 (PLIN1), a marker for triglyceride-rich adipocytes in 6 weeks old mice. (B) Quantitative data showing the number of cells stained positive for the expression of PLIN1 per myocardial section in the experimental and control groups at six weeks of age. (C) Transcript levels of selected markers of adipogenesis, quantified by RT-PCR, in the control and experimental groups. (D) Thin myocardial sections were stained for the expression of PLIN1 in the one-year-old Pdgfra-Cre:LmnaW/F and the control mice. (E) Quantitative data showing the number of PLIN1 expressing cells per section in one-year-old WT, Pdgfra-Cre, and Pdgfra-Cre:LmnaW/F mice. (F) Transcript levels of selected markers of adipogenesis in one-year-old WT, Pdgfra-Cre, and Pdgfra-Cre:LmnaW/F mice.
DEGs in the LMNA-deficient cardiac fibroblasts: Cardiac fibroblasts expressing PDGFRA but not the hematopoietic lineage markers were isolated by FACS from the WT and Pdgfra-Cre:LmnaF/F mice and used for RNA extraction and sequencing. Cardiac fibroblasts from the Pdgfra-Cre:LmnaW/F were not included as the phenotype in the latter group is largely similar to that in the Pdgfra-Cre:LmnaF/F mice, except that it evolves later in life. To reduce the confounding effects of overt cardiac dysfunction on gene expression, the RNA-Seq experiments were performed in the RNA extracted from cardiac fibroblasts isolated from the 3- to 4-week-old mice with preserved LVFS (LVEF: 55.5 ± 4.8 vs. 50.8% ± 7.4% in the WT and Pdgfra-Cre:LmnaF/F mice, respectively, P = 0.08). Approximately 20% of the lineage-negative sorted cells expressed PDGFRA in each genotype (WT: 20.9% ± 5.8% vs. Pdgfra-Cre:LmnaF/F 22.0 ± 6.1, P = 0.685), which were isolated and used in the RNA-sequencing [Figure 8A and B].

Figure 8. Differentially expressed genes, dysregulated transcriptional regulators, and biological pathways in the LMNA-deficient cardiac fibroblasts. (A) Fluorescence-activated cell sorting (FACS) panels showing the gating for IgG isotype and isolation of the cells expressing PDGFRA in the WT and Pdgfra-Cre:LmnaF/F mice. (B) Quantitative data showing the percentage of the non-myocyte cells isolated based on the signaling from an antibody against PDGFRA in the 6-week-old WT and Pdgfra-Cre:LmnaF/F mice. (C) Principle component analysis (PCA) of the cardiac fibroblast transcripts shows distinct separation between the WT and Pdgfra-Cre:LmnaF/F genotypes. (D) Volcano plot showing the down-regulated (blue), upregulated (red), and unchanged (black) genes in the Pdgfra-Cre:LmnaF/F as compared to the WT fibroblasts. (E) Heat map of the differentially expressed genes (DEGs) between the WT and Pdgfra-Cre:LmnaF/F fibroblasts. (F) The bar chart represents the predicted transcriptional regulators of the DEGs in Pdgfra-Cre:LmnaF/F fibroblasts. Red indicated predicted activation and blue predicted suppression. (G) The bar chart represents predicting growth and mitotic factors based on the DEGs in the Pdgfra-Cre:LmnaF/F fibroblasts. (H) Biological pathways predicted to be activated in the Pdgfra-Cre:LmnaF/F fibroblasts. (I) Biological pathways predicted to be suppressed in the Pdgfra-Cre:LmnaF/F fibroblasts. (J-M) Plots representing gene set enrichment analysis (GSEA) predicting activation of the TP53, TNFA/NFκB, and TGFβ1 and suppression of the cell cycle G2M pathways.
Principle Component Analysis (PCA) showed distinct genotype-dependent separation of the samples [Figure 8C]. Analysis of the transcript showed differential expression of 410 genes between the WT and Pdgfra-Cre:LmnaF/F mouse cardiac fibroblasts, which were comprised of 231 down-regulated and 179 upregulated genes [Figure 8D]. A heat map of the DEGs showed genotype-dependent expression of the dysregulated genes [Figure 8E]. Notable among the upregulated genes were Egr2 (4.2-fold), Ch25h
Increased expression levels of double-stranded DNA breaks (DSBs) and activation of the DDR Pathway: Given the predicted activation of the DDR pathway by the RNA-Seq data, expression, and levels of phospho-(p)H2AFX, a marker for the double-stranded DNA breaks (DSBs), were determined by immunofluorescence staining of the thin myocardial section, which showed an increased number of cell staining positive for pH2AFX in the Pdgfra-Cre:LmnaF/F hearts [Figure 9A and B]. To determine whether pH2AFX expression was induced in cardiac fibroblasts, isolated non-myocyte cells were co-stained for pH2AFX and PDGFRA, and the number of cells expressing both markers was determined, which showed a significant increase in cells isolated from the Pdgfra-Cre:LmnaF/F mouse hearts [Figure 9C and D]. To further corroborate the findings, expression levels of the pH2AFX and its upstream kinase ATM were determined by immunoblotting, which showed increased levels in the Pdgfra-Cre:LmnaF/F hearts [Figure 9E and F]. Finally, because DSBs are sensed by cytosolic DNA sensor CGAS protein, its expression level was assessed by immunoblotting, which also showed increased levels in the Pdgfra-Cre:LmnaF/F group [Figure 9E and F].

Figure 9. Activation of the DNA Damage Response (DDR) pathway. (A) Immunofluorescence panels showing expression of phospho-H2AFX, a marker for the double-stranded DNA breaks (DSBs) in the myocardial sections. (B) Quantitative data showing the percentage of nuclei staining positive for the expression of pH2AFX. (C) Immunofluorescence panels showing expression of phospho-H2AFX in isolated cardiac fibroblasts. Panels representing the expression of pH2AFX (green color), PDGFRA (red), and the overlay are presented along with enlarged inserts showing the expression of pH2AFX in the nuclei of isolated cardiac fibroblasts. (D) Graph depicting the percentage of the cells co-expressing PDGFRA and pH2AFX in the experimental and control groups. (E) Immunoblots showing expression of selected proteins involved in the DDR pathways, namely pH2AFX, total H2AFX, ATN, and CGAS, along with blot representing controls for the loading conditions, are shown in the WT, Pdgfra-Cre, Pdgfra-Cre:LmnaW/F, and Pdgfra-Cre:LmnaW/F mice. (F) Quantitative data corresponding to the blots shown in panel C.
Increased expression of the senescence markers: Given that the DDR pathway activates the cell senescence program by activating the TP53 and its bona fide target cell cycle inhibitor CDKN1A, expression levels of phosphoTP53 (residues Serine 18 and Serine 389) and CDKN1A were assessed by immunoblotting. Expression levels of pTP53 (Serine 18 and Serine 389) were increased in Pdgfra-Cre:LmnaF/F mouse hearts at six weeks of age, as were CDKN1A protein levels [Figure 10A and B]. In conjunction with the increased expression levels of cell cycle inhibitors, expression of the senescence-associated β-galactosidase was induced in Pdgfra-Cre:LmnaF/F mouse hearts [Figure 10C]. Likewise, expression levels of selected markers of senescence-associated secretory phenotype (SASP), namely CTGF (CCN2), and LGALS3, and as shown earlier TGFβ1, were increased in the hearts of six-week-old Pdgfra-Cre:LmnaF/F mice, whereas their expression was either lower or not detected in the WT, Pdgfra-Cre, and six-week-old heterozygous

Figure 10. Expression of senescence markers. (A) Immunoblots showing levels of phospho-TP53 S18 and S389 proteins as well as the bonafide downstream target of activation of TP53, namely CDKN1A, in the experimental groups. (B) Quantitative data representing the blots shown in panel A. (C) Thin myocardial sections were stained for the expression of senescence-associated β-galactosidase, detecting its expression in the heart of 6-week-old Pdgfra-Cre:LmnaF/F mice. (D) Immunoblots showing increased expression levels of CTGF (CCN2) and LGALS3, SASP markers, in the Pdgfra-Cre:LmnaF/F mouse hearts. Quantitative data are shown in panels (E and F). (G) Transcript levels of selected senescence-associated secretory phenotype (SASP), quantified by RT-PCR, showing increased transcript levels in the Pdgfra-Cre:LmnaF/F mouse hearts, as compared to other genotypes.
DISCUSSION
The findings highlight the contribution of cardiac fibroblasts to the pathogenesis of LMNA-associated DCM, as the deletion of the Lmna gene in fibroblasts leads to cardiac dilatation and dysfunction, cardiac arrhythmias, myocardial fibrosis, and apoptosis, a phenotype that resembles that of the DCM. The observed phenotype is not indistinct from DCM caused upon deletion of the Lmna gene in cardiac myocytes, albeit it is milder and occurs later in life[19,23]. Notably, systemic or myocyte-specific deletion of the Lmna gene leads to near-total mortality at three to four weeks of age, whereas the Pdgfra-Cre:LmnaF/F mice survive to eight weeks[19,23]. Similarly, cardiac dysfunction is milder and occurs later in the Pdgfra-Cre:LmnaF/F than
The expression of a DCM-like phenotype upon deletion of the Lmna gene in fibroblasts is in accord with the ubiquitous expression of the LMNA protein in various cell types, including cardiac fibroblasts[8]. Likewise, the observed phenotypic effects on cardiac function and arrhythmias likely reflect the cellular crosstalk between fibroblasts and myocytes, as well as the paracrine effects, including expression of the SASP. The latter mechanism is also in agreement with the paracrine effects resulting from the deletion of the placental growth factor in cardiac fibroblasts and the endothelial cells[29]. The findings also implicate increased DSBs, as evidenced by increased expression of the markers of the DSBs, activation of the DDR pathway, and increased expression of SASP, as the major mechanism in the pathogenesis of the phenotype. The findings, along with the data in cardiac myocyte-specific models of laminopathies, denote activation of the DDR pathway as a cell-autonomous pathogenic consequence of LMNA deficiency in the heart[19,23]. Collectively, the data underscore the role of the LMNA protein as a guardian of genomic integrity. Increased expression levels of the markers of DSBs and activation of the DDR pathway are also in accord with the findings upon deletion of another nuclear member protein, namely TMEM43. Together, the findings suggest a possible shared mechanism involving DNA damage in the pathogenesis of DCM in nuclear envelopathies[17].
The study has several limitations. The Lmna gene was deleted using the Pdgfra-Cre BAC transgenic deleter mice[15]. Although PDGFRA protein is abundantly expressed in cardiac fibroblasts, it is not an exclusive marker of cardiac fibroblasts, as it is also expressed in multiple mesenchymal tissues. No gross abnormalities were detected in other organs; however, the studies were primarily focused on evaluating the cardiac phenotype. Therefore, the possible presence of concomitant phenotypes in other organs cannot be excluded. The comprehensive metabolic panel did not show evidence of kidney or liver abnormalities or electrolyte disturbances. However, plasma glucose levels were reduced, and levels of the liver enzymes ALT and AST were modestly increased in the Pdgfra-Cre:LmnaF/F mice. The latter changes were likely reflective of the smaller body size observed in the Pdgfra-Cre:LmnaF/F mice. Overall, we surmise that the observed phenotype was cardiac-specific and likely the consequence of the deletion of the Lmna gene in cardiac fibroblasts but not due to abnormalities in other organs.
The PDGFRA is not expressed in mature cardiac myocytes; however, the locus is transcriptionally active in a small subset of cardiac myocytes during cardiogenesis[16]. In agreement with the previous data, using the Pdgfra-Cre mice led to the deletion of the Lmna gene in ~25% of cardiac myocytes in the Pdgfra-Cre:LmnaF/F mice, and correspondingly, the LMNA protein levels in cardiac myocytes were reduced by ~30%. Therefore, one might surmise that deletion of the Lmna gene in a small subset of cardiac myocytes also contributed to the expression of the DCM-like phenotype in the Pdgfra-Cre:LmnaF/F mice. The possibility cannot be excluded unambiguously. However, the heterozygous deletion of the Lmna gene in cardiac myocytes in the Myh6-Cre-LmnaW/F mice also leads to about 30% to 50% reduction in the LMNA levels in cardiac myocytes, does not induce an early phenotype, in contrast to the Pdgfra-Cre:LmnaF/F mice. Likewise, the
In conclusion, deficiency of the LMNA protein in cardiac fibroblasts contributes to the pathogenesis of the LMNA-associated DCM by activating the DDR pathway in response to increased DSBs and inducing fibroblast-senescence. The findings indicate a multi-cellular basis for the phenotype in LMNA-associated DCM. The findings of the present study, in conjunction with the previous data in myocyte-specific models, identify the DSBs as a major mechanism in the pathogenesis of LMNA-associated DCM and denote the LMNA protein as a guardian of genomic stability.
DECLARATIONS
Authors’ contributionsPerformed and analyzed the molecular biology and histology experiments as well as echocardiography and electrocardiography: Rouhi L, Auguste G, Zhou Q
Performed experiments on the RNA-Sequencing and secondary analysis of the data: Auguste G
Supervised and interpreted the echocardiographic data: Lombardi R
Performed some of the RT-PCR experiments: Olcum M
Assisted with genotyping and histological experiments and performed some of the RT-PCR experiments: Pourebrahim K
Analyzed the expression of the LMNA protein in the myocardium: Cheedipudi SM
Generated and maintained mouse colonies and genotyping by PCR: Asghar S
Provided a critical review of the manuscript: Hong K
Performed the primary and secondary analyses of the RNA-Seq data, including mapping of the reads, and edited the manuscript: Robertson MJ
Analyzed and interpreted the RNA-Seq data and edited the manuscript: Coarfa C
Analyzed the RNA sequencing data, interpreted the findings, and edited the manuscript: Gurha P
Conceived the idea, designed the experiments, interpreted the findings, and wrote the manuscript: Marian AJ
Availability of data and materialsRNA-Seq data have been submitted to GEO and available to the public upon release (GSE199078).
Financial support and sponsorshipDr. AJ Marian is supported by the National Heart, Lung, and Blood Institute (HL151737, HL132401). Dr. SM Cheedipudi is supported by Career Development Grant from American Heart Association (20CDA35310229).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateAnimal procedures were in full compliance with the NIH Guidelines for the Care and Use of Laboratory Animals and approved by the Animal Care and Use Committee of the University of Texas Health Science Center - Houston (AWC-21-0015).
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
Supplementary MaterialsREFERENCES
1. Emmons-Bell S, Johnson C, Roth G. Prevalence, incidence and survival of heart failure: a systematic review. Heart ;2022:heartjnl-2021.
2. James SL, Abate D, Abate KH, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018;392:1789-858.
3. Khush KK, Cherikh WS, Chambers DC, et al. The international thoracic organ transplant registry of the international society for heart and lung transplantation: thirty-sixth adult heart transplantation report - 2019; focus theme: donor and recipient size match. J Heart Lung Transplant 2019;38:1056-1066.
4. Hershberger RE, Cowan J, Jordan E, Kinnamon DD. The complex and diverse genetic architecture of dilated cardiomyopathy. Circ Res 2021;128:1514-1532.
5. McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res 2017;121:731-748.
6. Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med 1999;341:1715-1724.
7. Taylor MR, Fain PR, Sinagra G, et al. Familial dilated cardiomyopathy registry research g. natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol 2003;41:771-780.
8. Rober RA, Weber K, Osborn M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development 1989;105:365-378.
9. Schreiber KH, Kennedy BK. When lamins go bad: nuclear structure and disease. Cell 2013;152:1365-1375.
10. Vidak S, Foisner R. Molecular insights into the premature aging disease progeria. Histochem Cell Biol 2016;145:401-417.
12. Anselme F, Moubarak G, Savoure A, et al. Implantable cardioverter-defibrillators in lamin A/C mutation carriers with cardiac conduction disorders. Heart Rhythm 2013;10:1492-1498.
13. Van Berlo JH, de Voogt WG, van der Kooi AJ, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med (Berl) 2005;83:79-83.
14. Kim Y, Zheng Y. Generation and characterization of a conditional deletion allele for Lmna in mice. Biochem Biophys Res Commun 2013;440:8-13.
15. Roesch K, Jadhav AP, Trimarchi JM, et al. The transcriptome of retinal Muller glial cells. J Comp Neurol 2008;509:225-238.
16. Lombardi R, Chen SN, Ruggiero A, et al. Cardiac Fibro-adipocyte progenitors express desmosome proteins and preferentially differentiate to adipocytes upon deletion of the desmoplakin gene. Circ Res 2016;119:41-54.
17. Rouhi L, Cheedipudi SM, Chen SN, et al. Haploinsufficiency of Tmem43 in cardiac myocytes activates the DNA damage response pathway leading to a late-onset senescence-associated pro-fibrotic cardiomyopathy. Cardiovasc Res 2021;117:2377-2394.
18. Cheedipudi SM, Hu J, Fan S, et al. Exercise restores dysregulated gene expression in a mouse model of arrhythmogenic cardiomyopathy. Cardiovasc Res 2020;116:1199-1213.
19. Auguste G, Rouhi L, Matkovich SJ, et al. BET bromodomain inhibition attenuates cardiac phenotype in myocyte-specific lamin A/C-deficient mice. J Clin Invest 2020;130:4740-4758.
20. Devereux RB, Alonso DR, Lutas EM, et al. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol 1986;57:450-458.
21. Chen SN, Lombardi R, Karmouch J, et al. DNA Damage Response/TP53 Pathway Is Activated and Contributes to the Pathogenesis of Dilated Cardiomyopathy Associated With LMNA (Lamin A/C) Mutations. Circ Res 2019;124:856-873.
22. Olcum M, Cheedipudi SM, Rouhi L, et al. The WNT/beta-catenin pathway regulates expression of the genes involved in cell cycle progression and mitochondrial oxidative phosphorylation in the postmitotic cardiac myocytes. J Cardiovasc Aging 2022;2:15.
23. Auguste G, Gurha P, Lombardi R, Coarfa C, Willerson JT, Marian AJ. Suppression of activated FOXO transcription factors in the heart prolongs survival in a mouse model of laminopathies. Circ Res 2018;122:678-692.
24. Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 2019;37:907-915.
25. Liao Y, Smyth GK, Shi W. Featurecounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30:923-930.
26. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550.
27. Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med 1990;9:811-818.
28. Rouhi L, Fan S, Cheedipudi SM, et al. Effects of tamoxifen inducible MerCreMer on gene expression in cardiac myocytes in mice. J Cardiovasc Aging 2022;2:8.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Rouhi L, Auguste G, Zhou Q, Lombardi R, Olcum M, Pourebrahim K, Cheedipudi SM, Asghar S, Hong K, Robertson MJ, Coarfa C, Gurha P, Marian AJ. Deletion of the Lmna gene in fibroblasts causes senescence-associated dilated cardiomyopathy by activating the double-stranded DNA damage response and induction of senescence-associated secretory phenotype. J Cardiovasc Aging 2022;2:30. http://dx.doi.org/10.20517/jca.2022.14
AMA Style
Rouhi L, Auguste G, Zhou Q, Lombardi R, Olcum M, Pourebrahim K, Cheedipudi SM, Asghar S, Hong K, Robertson MJ, Coarfa C, Gurha P, Marian AJ. Deletion of the Lmna gene in fibroblasts causes senescence-associated dilated cardiomyopathy by activating the double-stranded DNA damage response and induction of senescence-associated secretory phenotype. The Journal of Cardiovascular Aging. 2022; 2(3): 30. http://dx.doi.org/10.20517/jca.2022.14
Chicago/Turabian Style
Rouhi, Leila, Gaelle Auguste, Qiong Zhou, Raffaella Lombardi, Melis Olcum, Kimia Pourebrahim, Sirisha M. Cheedipudi, Saman Asghar, Kui Hong, Matthew J. Robertson, Cristian Coarfa, Priyatansh Gurha, Ali J. Marian. 2022. "Deletion of the Lmna gene in fibroblasts causes senescence-associated dilated cardiomyopathy by activating the double-stranded DNA damage response and induction of senescence-associated secretory phenotype" The Journal of Cardiovascular Aging. 2, no.3: 30. http://dx.doi.org/10.20517/jca.2022.14
ACS Style
Rouhi, L.; Auguste G.; Zhou Q.; Lombardi R.; Olcum M.; Pourebrahim K.; Cheedipudi SM.; Asghar S.; Hong K.; Robertson MJ.; Coarfa C.; Gurha P.; Marian AJ. Deletion of the Lmna gene in fibroblasts causes senescence-associated dilated cardiomyopathy by activating the double-stranded DNA damage response and induction of senescence-associated secretory phenotype. J. Cardiovasc. Aging. 2022, 2, 30. http://dx.doi.org/10.20517/jca.2022.14
About This Article
Copyright

Data & Comments
Data


 Cite This Article 22 clicks
Cite This Article 22 clicks

 Like This Article 13
likes
Like This Article 13
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.