
Dysfunctional mitochondria elicit bioenergetic decline in the aged heart
 0
0
Abstract
Aging represents a complex biological progression affecting the entire body, marked by a gradual decline in tissue function, rendering organs more susceptible to stress and diseases. The human heart holds significant importance in this context, as its aging process poses life-threatening risks. It entails macroscopic morphological shifts and biochemical changes that collectively contribute to diminished cardiac function. Among the numerous pivotal factors in aging, mitochondria play a critical role, intersecting with various molecular pathways and housing several aging-related agents. In this comprehensive review, we provide an updated overview of the functional role of mitochondria in cardiac aging.
Keywords
INTRODUCTION
Aging is known to induce alterations in the heart, irrespective of specific cardiovascular risk factors like diabetes or hypertension, resulting in cardiac remodeling and functional changes.
CLINICAL OBSERVATIONS
Standard echocardiography reveals modifications in cardiac dimensions and wall thickness. The prevalence of left ventricular hypertrophy (LVH) increases with age, even in the absence of hypertension[1-5]. Diastolic dysfunction is a prevailing feature, evidenced by compromised LV filling in early diastolic phase (peak E wave), potentially due to fibrosis and reduced ventricular elasticity[6]. Atrial contraction (A wave) increases with age, contributing to atrial hypertrophy and heightened atrial fibrillation risk[7-10]. The E/A ratio, reflecting diastolic LV filling dynamics, declines significantly with age, indicating an increased reliance on late diastolic filling, suggestive of diastolic dysfunction[9,11]. Torsion, a characteristic rotational deformation during systole, increases with age, potentially indicating subendocardial dysfunction due to fibrosis[12,13].
NEUROHORMONAL SYSTEMS AND CARDIAC AGING
The adrenergic system and the renin-angiotensin-aldosterone system (RAAS) play decisive roles in cardiac remodeling during aging. Cardiac aging involves progressive collagen deposition, leading to interstitial fibrosis and altered cardiac elasticity, predominantly affecting diastole over systole. Elevated intracardiac concentrations of Angiotensin II (Ang II) accompany aging, substantially contributing to functional, molecular, and structural modifications consistent with Ang II effects[14,15]. In fact, inhibiting Ang II signaling by enalapril or losartan has been shown to extend lifespan and delay age-related cardiovascular pathologies[16-19].
Cardiac aging also features a substantial depletion of autonomic fibers and reduced cardiac and circulating levels of brain-derived neurotrophic factor (BDNF)[20]. The latter rules basal myocardial contractility/relaxation[21] and metabolism[22].
The adrenergic system, particularly through β adrenergic receptors (βARs), is intimately linked to cardiac aging, a process marked by a decline in βAR sensitivity and density, which is fairly consistent across species[9,23,24].
Overall, aging exhibits an uncontrolled activation of the adrenergic system, impacting βARs, their subtypes
The adrenergic system is implicated in metabolic alterations, especially in heart failure[32,33]. Cardiac bioenergetic changes with age, as evidenced, for instance, by alterations in the creatine kinase enzyme system and by increased catecholamines, eventually affecting mitochondrial function[34-36]. Indeed, chronic
MITOCHONDRIAL DYSFUNCTION IN CARDIAC AGING
Mitochondria, often referred to as cell powerhouses, generate adenosine triphosphate (ATP) through oxidative phosphorylation, a meticulously regulated process for maintaining cellular energy balance[41,42]. The mitochondrial electron transport chain (ETC) contributes 80%-90% of ATP in most mammalian tissues, making mitochondrial dysfunction detrimental due to reduced ATP production, indispensable for biological functions[43,44]. Aging leads to alterations in ETC components, contributing to a number of age-related conditions[45-48]. Hence, one of the consequences of the aging process is the drop in the efficiency of this energy production mechanism, leading to a decline in ATP production and subsequent cellular energy deficits[49,50]. In fact, dysfunctional mitochondria are increasingly associated with aged cardiovascular tissues.
Proper substrate utilization is imperative for the myocardium to fulfill its function, primarily relying on ATP (re-)synthesis through fatty acid oxidation within mitochondria[51]. The myocyte employs additional pathways, such as glycolysis, creatine kinase, and adenylate kinase, in response to high ATP demand. During increased work, glycogen, glucose, and phosphocreatine are utilized to meet ATP demand, maintaining constant ATP levels. The efficiency of ATP production varies depending on substrate oxidation, with fatty acid oxidation producing more ATP on a molar basis[52]. This metabolic plasticity diminishes in chronic pathologies like congestive heart failure, impacting myocardial oxygen efficiency and causing intracellular ATP depletion.
Spare respiratory capacity, the ability to increase ATP production during heightened demand or reduced fuel supply, is crucial for cellular function and survival. Mitochondrial plasticity involves the efficiency of mitochondrial coupling and provides increased respiratory capacity under stress conditions. This phenomenon is particularly significant in ischemic injury and situations of augmented energy demand such as sepsis, endurance exercise, trauma, or heart failure[53]. Mitochondrial plasticity is decreased in patients with insulin resistance and in elderly individuals, impacting cell function[54]. In the heart, aging leads to a decline in mitochondrial oxidative phosphorylation function, especially in state 3 respiration, related to electron transport complexes I and IV[55]. Increased electron leakage and mitochondrial ROS production contribute to oxidative damage, particularly affecting intrafibrillar mitochondria. Deficient mitochondrial energetics, altered Ca2+ homeostasis, and excessive ROS generation contribute to reduced stress adaptability and augmented vulnerability to disease in the aged myocardium[56,57]. Interconnected communication between the sarcoplasmic reticulum and mitochondria supports local signal transduction and ionic exchange[58-62]. Mitochondrial Ca2+ uptake plays fundamental roles in energy supply-demand matching and antioxidative defenses. Aging negatively impacts mitochondria-SR communication, potentially involving an impaired Ca2+ transmission and decreased physical interaction between ryanodine receptors (RyR) and mitochondrial VDAC (voltage-dependent anion channel)[63,64].
Insulin receptor signaling is central in mediating processes boosting glucose uptake in cardiomyocytes, and cardiac insulin resistance is known to contribute to ventricular dysfunction by favoring fatty acid utilization[65]. Insulin resistance is associated with metabolic inefficiency and impaired mitochondrial fitness[66,67], leading to contractile dysfunction. Mitochondrial abnormalities, reduced expression of oxidative phosphorylation regulators, increased reactive oxygen species (ROS), and compromised mitochondrial biogenesis and myocyte energetics are observed in senescent hearts. Molecular modifications characteristic of heart failure, such as shifts in membrane receptor signaling, survival-kinase signal transduction, and mitochondrial dysfunction/cell death, are also observed in the senescent heart[68].
MITOCHONDRIAL OXIDATIVE STRESS IN AGING
In the aging heart, mitochondria become a significant source of ROS. Their excessive production leads to a consequent imbalance with the vasoprotective bioavailability of nitric oxide[69]. ROS, also known as oxygen-containing reactive chemical species, represent a variety of oxidizing chemical compounds ranging from the highly reactive hydroxyl group to hydrogen peroxide[70].
ROS production leads to lipid peroxidation and the onset of DNA damage, which contribute to accelerating the processes linked to cardiac aging. Within intact cardiomyocytes, major sources of ROS commonly derive from the mitochondrial OXPHOS complexes I (i.e., NADH dehydrogenase) and III (i.e., cytochrome bc1 complex), which are part of the electron transport chain (ETC)[71].
In the ETC, NADH and FADH2 supply electrons to complex I and complex II (i.e., succinate dehydrogenase). These electrons are subsequently transmitted to complex III via ubiquinone (i.e., CoQ, coenzyme Q) and then progress through complex IV (i.e., cytochrome c-oxidase) until reaching molecular O2, which represents the final acceptor[72]. The sources of origin of ROS are multiple, but their main production is attributed to reverse electron transport via complex I through means of high proton driving force or low CoQ availability[73]. Furthermore, ROS can emerge from single electrons escaping from the ETC in an inactive process called electron scattering[74]. The escape of these electrons leads them to interact with O2, reducing it to generate a very unstable superoxide anion (O2- or O2·-)[75]. These cytotoxic oxygen intermediates are metabolized by glutathione peroxidase (GPX), catalase (Cat), and superoxide dismutase (SOD)[76-78], antioxidant systems that are extremely efficient in healthy cardiomyocytes. To counteract this ROS production, mitochondria employ this network of ROS-scavenging enzyme systems, which attempt to mitigate the excessive stress produced[79-81]. SOD catalyzes the conversion of O2- into hydrogen peroxide
Mitochondria engaged in respiration continually generate H2O2. If the production surpasses scavenging capacity, H2O2 emission takes place, posing a threat to cellular functions[71,85-88]. Peroxiredoxin-3, a mitochondrial peroxidase, has been shown to mitigate H2O2 by converting it to H2O, utilizing reducing equivalents from NADPH provided by thioredoxin-2 and thioredoxin reductase-2[89].
The physiologic pattern of ROS production becomes exacerbated in aging because of detrimental attacks within the cardiomyocyte over time[90-93]. It also determines the accretion of dysfunctional organelles and damaged proteins, which contribute to increasing the incidence of many age-related cardiovascular diseases[94,95].
Over the years, several studies have been conducted on a murine model overexpressing human antioxidant catalase (mCAT)[96,97]. This overexpression increased lifespan due to decreased oxidative stress and led to phenotypes with reduced cardiac aging, substantiated by improved contractile function and mitigated hypertrophy[98]. The mCAT mouse models also displayed attenuated mitochondrial H2O2 toxicity, decreased oxidative DNA damage, and decreased accumulation of mutations in mitochondrial DNA (mtDNA)[99].
Cardiomyocytes are long-lived postmitotic cells that can live for many years: they have a limited capacity for regeneration and a rare propensity for malignant transformation[100,101]. Intriguing theories related to mitochondrial aging in the heart are related to the high mutation rate and limited repair capacity of mtDNA, compromising the integrity of the mitochondrial genome[102,103]. As a consequence, in many cardiac cells undergoing the aging process, mitochondrial functions are impaired and their ability to generate energy is progressively lost, counterbalanced by increased generation of ROS[104].
The presence of deletions and point mutations in mtDNA has been reported to increase with age in various tissues including the myocardium[105]. A previous study has demonstrated increased levels of H2O2 in subsarcolemmal mitochondria (SSM) and increased levels of oxidative stress in interfibrillar mitochondria (IFM) in isolated hearts of aged rats compared to young ones[106].
MITOCHONDRIA AND INFLAMMAGING
By activating different redox signaling pathways, mitochondrial ROS are known to contribute to chronic vascular inflammation in aging[107]. Chronic inflammatory processes are recognized as contributing factors to age-related cardiovascular disorders such as vascular cognitive impairment, heart failure, peripheral artery disease, and stroke[108]. For instance, when inflammation occurs, there are many pro-inflammatory changes in the endothelial phenotype. They include an increase in cell adhesion molecules, augmented endothelium-leukocyte interactions, and changes in the secretion of pro-inflammatory molecules. Previous investigations have shown that advanced age induces modifications in cytokine profiles and triggers the expression of pro-inflammatory genes in cardiac muscle, in the perivascular adipose tissue, and in the walls of large arteries[109,110]. The activation of t NF-κB plays an essential role in pro-inflammatory modifications and endothelial activation in the vasculature associated with aging both in humans[111] and in laboratory mouse models[107].
Emerging evidence indicates that mitochondria-associated factors may activate a number of innate immune mechanisms that modulate host defenses, such as interferon (IFN)-dependent antiviral responses and inflammasome-dependent pro-inflammatory cytokines[112].
AUTOPHAGY AND AGING
Autophagy, a fundamental process in organelle and protein degradation, plays a key role in programmed cell death[113-116]. Three main types of autophagy have been described: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA)[117-119]. Macroautophagy involves the sequestration of portions of cytoplasm within autophagosomes, double-membrane-enclosed vacuoles, which then fuse with lysosomes for subsequent degradation[120]. Microautophagy, on the other hand, does not require sequestration; small portions of cytoplasm enter lysosomes via invaginations within the limiting membrane[121].
CMA does not involve vesicular traffic. In CMA, cytosolic proteins with specific peptide sequence motifs (KFERQ) are recognized by molecular chaperones. This substrate-chaperone complex then binds to Lamp2a (lysosome-associated membrane protein type 2a), a receptor on the lysosomal membrane, facilitating the entry of proteins into the lysosomal lumen[121,122].
CARDIOPROTECTIVE EFFECTS OF AUTOPHAGY
Numerous studies have demonstrated that inducing autophagy in the heart has cardioprotective effects[123-129]. Additionally, autophagy tends to decrease during aging, and interventions that enhance autophagy have shown promise in reducing age-related cardiac disorders and extending lifespan[130-132]. However, excessive autophagy might prove harmful, as it may lead to a disproportionate degradation of essential cellular components, ultimately resulting in cell death[133,134].
MAIN INTRACELLULAR MECHANISMS UNDERLYING AUTOPHAGY
Specifically, various intracellular mechanisms play a critical role in either inhibiting or promoting autophagy, a process intimately associated with aging[114,135]. For instance, mTOR acts as a sensor[136] for energy metabolism and nutrient availability, negatively regulating autophagy: mTOR forms two complexes, mTORC1 and mTORC2; mTORC1, linked to aging, inhibits autophagy via phosphorylation of the
SIRT1, a member of the Sirtuin family, regulates intracellular bioenergetics. It deacetylates FoxO1, promoting autophagy, and also deacetylates ATG proteins. SIRT1 is cardioprotective, but chronic elevated levels can be detrimental, leading to oxidative stress, apoptosis, and cardiomyopathy[148-152]. Proper regulation of SIRT1 is necessary for maintaining cardiac function[150,153,154]. The Sirtuin family is also essential for the regulation of the mitochondrial unfolded protein response (mtUPR), one of the first stress-protective responses started by mitochondrial dysfunction[155], which clears or repairs misfolded proteins in order to mitigate the damage[156-158].
Equally important, the induction of endoplasmic reticulum stress (ER stress) has been shown to damage the ETC in aged cardiac mitochondria, in a process regulated by mitochondria-localized calpain 1 and
Eventually, oxidative damage towards the cardiovascular system leads to functional failure of the mitochondrion[160-164]. Hence arises the necessity for the cell to eliminate these dysfunctional organelles in order to preserve cellular homeostasis[165,166].
RECEPTOR-MEDIATED MITOPHAGY
Damaged mitochondria are selected for degradation via a specific form of autophagy (mitophagy)[141,167]. Receptor-mediated mitophagy relies on receptors located on the outer mitochondrial membrane (OMM). These receptors, including Bcl2/adenovirus E1B 19-kDa interacting protein 3 (BNIP3), Bcl-2-like protein 13 (Bcl2L13/Bcl-Rambo), NIX/BNIP3L, FUN14 domain-containing 1 (FUNDC1), and FK506-binding protein (FKBP8), are anchored to the OMM and directly bind to LC3, guiding the targeted organelle to the autophagosome without ubiquitination[168-170]. Limited information exists on the exact role of mitophagy players during aging, but it is known that BNIP3 is upregulated in aged hearts[171]. Notably, NIX and BNIP3 may transition to maladaptive inducers of cell death in age-induced cardiac stress, deviating from their role in promoting mitochondrial quality control[172,173].
PARKIN AND CARDIAC AGING
The PINK1-Parkin mitophagy pathway (PTEN-induced putative kinase protein 1, Parkinson Protein 2) involves targeted degradation of damaged mitochondria[174]. PINK1, under normal conditions, translocates and is degraded within the mitochondria. PINK1 and Parkin target and degrade damaged and poorly functioning mitochondria[175,176]: following oxidative stress-induced membrane depolarization, PINK1 accumulates, recruiting Parkin, which ubiquitinates damaged mitochondria[177,178]. Autophagosomes enclose these mitochondria, fusing with lysosomes to initiate degradation. Loss of Parkin function hinders effective clearance, potentially triggering further mitochondrial fission[175]. Many investigations focused on vascular aging have confirmed the role of mitophagy mediated by PINK1 and Parkin. A reduction in Parkin expression was detected in the aorta of aged mice and was then correlated with increased superoxide production and aortic stiffness[179]. Theablation of Parkin in aged mice was shown to cause accumulation of abnormal mitochondria in the heart, and interestingly, mice lacking Parkin showed enhanced aging compared to controls in which the protein was normally expressed[180]. On the contrary, overexpression of Parkin in cardiomyocytes ameliorated mitochondrial health, slowing down cardiac aging[181].
Over the years, clinical investigations have confirmed the preclinical hypotheses demonstrating reduced cardiac expression of PINK1 in elderly patients with heart failure compared with healthy individuals[182].
AGING AND MITOCHONDRIAL DYNAMICS
Mitochondrial dynamics, including fusion and fission events, are integral to maintaining mitochondrial function. Imbalances in these dynamic processes have also been implicated in cardiovascular aging[100,168,183,184]. Excessive fission results in fragmented mitochondria, promoting cellular dysfunction, while impaired fusion compromises mitochondrial networking and bioenergetic efficiency[183,185].
Specific dynamin-related GTPases play functional roles in the mitochondrial fusion process: optic atrophy 1 (OPA1) for inner membrane fusion and mitofusins (MFN1 and MFN2) for outer mitochondrial membrane (OMM) fusion[186-188].
Fission leads to the division of mitochondria into two separate organelles. Drp1 (dynamin-related protein 1) is the master regulator of mitochondrial fission. Upon activation, Drp1 translocates from the cytosol to the mitochondria[189-191] and then it forms a coiled ring with an internal diameter of ~20 nm[192]. All the morphological alterations caused by inadequate fission and fusion processes play a role in cardiac aging, resulting in fibrosis, diastolic dysfunction, and ventricular hypertrophy. A reduced expression of MFN1-MFN2 has been reported in 25-month-old rats, with an increase in Opa1 and Drp1 in 36-month-old rats[193]. A shift towards mitochondrial fusion exacerbates cellular senescence, as highlighted by studies of senescence promoting Fis1 knockdown[194].
Furthermore, alterations in mitochondrial fusion accelerate cardiac aging. Opa1+/- mice showed fragmented mitochondria and an impaired myocardial function[195]. Similarly, Mfn2-deficient mice exhibited impaired left ventricular function by 17 months[196]. A balance between the mechanisms of mitochondrial fusion and fission would allow the cardiac tissue to be preserved from the onset of age-related anomalies[197-200].
CONCLUSIONS
Unraveling the molecular intricacies of the relationship linking mitochondria to myocardial health provides a foundation for developing targeted interventions to mitigate age-related cardiovascular decline [Figure 1]. As research in this field continues to evolve, the prospect of therapeutic strategies aimed at preserving mitochondrial fitness and promoting cardiovascular health in aging populations becomes increasingly promising. Therefore, strategies aimed at improving mitochondrial function in older patients may be cardio-protective.
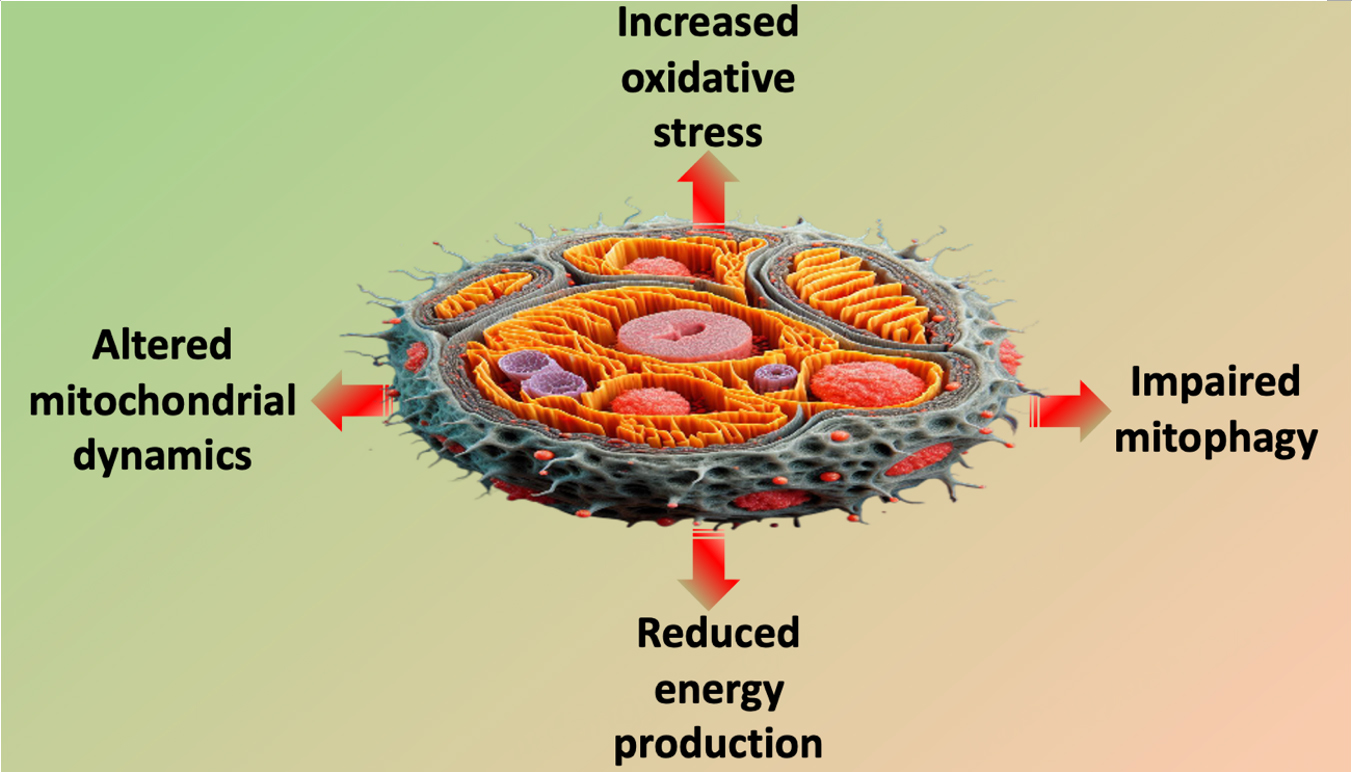
Figure 1. Main mitochondrial alterations in cardiovascular aging.
DECLARATIONS
Authors’ contributions
Drafted the manuscript: Mone P, Agyapong ED, Jankauskas SS, Varzideh F
Edited and revised the manuscript: Morciano G, De Luca A, Pinton P, Santulli G
Availability of data and materials
Not applicable.
Financial support and sponsorship
The Santulli’s Lab is currently supported in part by the National Institutes of Health (NIH): National Heart, Lung, and Blood Institute (NHLBI: R01-HL164772, R01-HL159062, R01-HL146691, T32-HL144456), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK: R01-DK123259, R01-DK033823), National Center for Advancing Translational Sciences (NCATS: UL1-TR002556-06, UM1-TR004400) to G.S., by the Diabetes Action Research and Education Foundation (to G.S.), and by the Monique Weill-Caulier and Irma T. Hirschl Trusts (to G.S.). This publication was also produced with the co-funding of the European Union - Next Generation EU, in the context of The National Recovery and Resilience Plan, PE8 Investment, Project Age-It (Ageing Well in an Ageing Society), to P.M. The Santulli lab is also supported in part by the American Heart Association (AHA-21836407 to SSJ, AHA-22915561 and AHA-241195524 to FV).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024.
REFERENCES
1. Huang M, Li J, Zhao X, Chen S, Li X, Jiang W. Relationship between vascular ageing and left ventricular geometry in patients with newly diagnosed primary aldosteronism. Front Endocrinol 2022;13:961882.
2. Pelà G, Tagliaferri S, Perrino F, et al. Determinants of cardiac structure in frail and sarcopenic elderly adults. Exp Gerontol 2021;150:111351.
3. Lembo M, Trimarco V, Izzo R, et al. Achieving a systolic blood pressure below 130 mmHg reduces the incidence of cardiovascular events in hypertensive patients with echocardiographic left ventricular hypertrophy. J Pharmacol Exp Ther 2023:388.
4. Maimaitiaili R, Teliewubai J, Zhao S, et al. Relationship between vascular aging and left ventricular concentric geometry in community-dwelling elderly: the northern shanghai study. Clin Interv Aging 2020;15:853-63.
5. Prasad A, Popovic ZB, Arbab-Zadeh A, et al. The effects of aging and physical activity on Doppler measures of diastolic function. Am J Cardiol 2007;99:1629-36.
6. Arbab-Zadeh A, Dijk E, Prasad A, et al. Effect of aging and physical activity on left ventricular compliance. Circulation 2004;110:1799-805.
7. van Ommen AMLN, Canto ED, Cramer MJ, Rutten FH, Onland-Moret NC, Ruijter HMD. Diastolic dysfunction and sex-specific progression to HFpEF: current gaps in knowledge and future directions. BMC Med 2022;20:496.
8. Nishimura T, Senoo K, Makino M, et al. Prediction model for the new onset of atrial fibrillation combining features of 24-hour Holter electrocardiogram with 12-lead electrocardiogram. Int J Cardiol Heart Vasc 2023;47:101245.
9. Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part III: cellular and molecular clues to heart and arterial aging. Circulation 2003;107:490-7.
10. Santulli G, D’ascia SL, D’ascia C. Development of atrial fibrillation in recipients of cardiac resynchronization therapy: the role of atrial reverse remodelling. Can J Cardiol 2012;28:245.e17-8.
11. Bursi F, Weston SA, Redfield MM, et al. Systolic and diastolic heart failure in the community. JAMA 2006;296:2209-16.
12. Fleg JL. Diagnostic and prognostic value of stress testing in older persons. J Am Geriatr Soc 1995;43:190-4.
13. Correia LC, Lakatta EG, O’Connor FC, et al. Attenuated cardiovascular reserve during prolonged submaximal cycle exercise in healthy older subjects. J Am Coll Cardiol 2002;40:1290-7.
14. Groban L, Pailes NA, Bennett CD, et al. Growth hormone replacement attenuates diastolic dysfunction and cardiac angiotensin II expression in senescent rats. J Gerontol 2006;61:28-35.
15. Dai DF, Santana LF, Vermulst M, et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 2009;119:2789-97.
16. Basso N, Cini R, Pietrelli A, Ferder L, Terragno NA, Inserra F. Protective effect of long-term angiotensin II inhibition. Am J Physiol 2007;293:H1351-8.
17. Benigni A, Corna D, Zoja C, et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. J Clin Invest 2009;119:524-30.
18. Santulli G, Ciccarelli M, Trimarco B, Iaccarino G. Physical activity ameliorates cardiovascular health in elderly subjects: the functional role of the β adrenergic system. Front Physiol 2013;4:209.
19. Stein M, Boulaksil M, Jansen JA, et al. Reduction of fibrosis-related arrhythmias by chronic renin-angiotensin-aldosterone system inhibitors in an aged mouse model. Am J Physiol Heart Circ Physiol 2010;299:H310-21.
20. Elia A, Cannavo A, Gambino G, et al. Aging is associated with cardiac autonomic nerve fiber depletion and reduced cardiac and circulating BDNF levels. J Geriatr Cardiol 2021;18:549-59.
21. Feng N, Huke S, Zhu G, et al. Constitutive BDNF/TrkB signaling is required for normal cardiac contraction and relaxation. Proc Natl Acad Sci USA 2015;112:1880-5.
22. Yang X, Zhang M, Xie B, et al. Myocardial brain-derived neurotrophic factor regulates cardiac bioenergetics through the transcription factor Yin Yang 1. Cardiovasc Res 2023;119:571-86.
23. White M, Roden R, Minobe W, et al. Age-related changes in beta-adrenergic neuroeffector systems in the human heart. Circulation 1994;90:1225-38.
24. Santulli G, Iaccarino G. Adrenergic signaling in heart failure and cardiovascular aging. Maturitas 2016;93:65-72.
25. Santulli G, Iaccarino G. Pinpointing beta adrenergic receptor in ageing pathophysiology: victim or executioner? Evidence from crime scenes. Immun Ageing 2013;10:10.
26. Perino A, Ghigo A, Ferrero E, et al. Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110γ. Mol Cell 2011;42:84-95.
27. Nikolaev VO, Moshkov A, Lyon AR, et al. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010;327:1653-7.
28. Lyon AR, Nikolaev VO, Miragoli M, et al. Plasticity of surface structures and β2-adrenergic receptor localization in failing ventricular cardiomyocytes during recovery from heart failure. Circ Heart Fail 2012;5:357-65.
29. Okumura S, Takagi G, Kawabe J, et al. Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc Natl Acad Sci USA 2003;100:9986-90.
30. Okumura S, Vatner DE, Kurotani R, et al. Disruption of type 5 adenylyl cyclase enhances desensitization of cyclic adenosine monophosphate signal and increases Akt signal with chronic catecholamine stress. Circulation 2007;116:1776-83.
31. Yan L, Vatner DE, O’Connor, et al. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell 2007;130:247-58.
32. Ciccarelli M, Chuprun JK, Rengo G, et al. G protein-coupled receptor kinase 2 activity impairs cardiac glucose uptake and promotes insulin resistance after myocardial ischemia. Circulation 2011;123:1953-62.
33. Ciccarelli M, Santulli G, Pascale V, Trimarco B, Iaccarino G. Adrenergic receptors and metabolism: role in development of cardiovascular disease. Front Physiol 2013;4:265.
35. Maslov MY, Chacko VP, Stuber M, et al. Altered high-energy phosphate metabolism predicts contractile dysfunction and subsequent ventricular remodeling in pressure-overload hypertrophy mice. Am J Physiol 2007;292:H387-91.
36. Schocke MF, Metzler B, Wolf C, et al. Impact of aging on cardiac high-energy phosphate metabolism determined by phosphorus-31 2-dimensional chemical shift imaging (31P 2D CSI). Magn Reson Imaging 2003;21:553-9.
37. Remondino A, Kwon SH, Communal C, et al. β-adrenergic receptor-stimulated apoptosis in cardiac myocytes is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of the mitochondrial pathway. Circ Res 2003;92:136-8.
38. No MH, Choi Y, Cho J, et al. Aging promotes mitochondria-mediated apoptosis in rat hearts. Life 2020;10:178.
39. Assali EA, Jones AE, Veliova M, et al. NCLX prevents cell death during adrenergic activation of the brown adipose tissue. Nat Commun 2020;11:3347.
40. Santulli G, Kansakar U, Varzideh F, Mone P, Jankauskas SS, Lombardi A. Functional role of taurine in aging and cardiovascular health: an updated overview. Nutrients 2023;15:4236.
41. Giorgi C, Marchi S, Pinton P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol 2018;19:713-30.
42. Pekson R, Liang FG, Axelrod JL, et al. The mitochondrial ATP synthase is a negative regulator of the mitochondrial permeability transition pore. Proc Natl Acad Sci USA 2023;120:e2303713120.
43. Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders - a step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis 2017;1863:1066-77.
44. Zorova LD, Popkov VA, Plotnikov EY, et al. Mitochondrial membrane potential. Anal Biochem 2018;552:50-9.
45. Jedlička J, Tůma Z, Razak K, et al. Impact of aging on mitochondrial respiration in various organs. Physiol Res 2022;71:S227-36.
46. Strait JB, Lakatta EG. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail Clin 2012;8:143-64.
47. Ribeiro ASF, Zerolo BE, López-Espuela F, Sánchez R, Fernandes VS. Cardiac system during the aging process. Aging Dis 2023;14:1105-22.
48. Rysz J, Franczyk B, Rysz-Górzyńska M, Gluba-Brzózka A. Ageing, age-related cardiovascular risk and the beneficial role of natural components intake. Int J Mol Sci 2021;23:183.
49. Tocchi A, Quarles EK, Basisty N, Gitari L, Rabinovitch PS. Mitochondrial dysfunction in cardiac aging. Biochim Biophys Acta 2015;1847:1424-33.
50. Nanadikar MS, Vergel Leon AM, Guo J, et al. IDH3γ functions as a redox switch regulating mitochondrial energy metabolism and contractility in the heart. Nat Commun 2023;14:2123.
51. Shi X, Qiu H. New insights into energy substrate utilization and metabolic remodeling in cardiac physiological adaption. Front Physiol 2022;13:831829.
52. Gambardella J, Jankauskas SS, Kansakar U, et al. Ketone bodies rescue mitochondrial dysfunction via epigenetic remodeling. JACC Basic Transl Sci 2023;8:1123-37.
53. Liu Q, Pan S, Li P, Dixon RAF. Hippo pathway inhibition promotes metabolic adaptability and antioxidant response in myoblasts. Sci Rep 2023;13:2232.
54. Mone P, Morgante M, Pansini A, et al. Effects of insulin resistance on mitochondrial (dys)function. Atherosclerosis 2022;341:52-4.
55. Kane MS, Benavides GA, Osuma E, et al. The interplay between sex, time of day, fasting status, and their impact on cardiac mitochondrial structure, function, and dynamics. Sci Rep 2023;13:21638.
56. Hernandez-Resendiz S, Prakash A, Loo SJ, et al. Targeting mitochondrial shape: at the heart of cardioprotection. Basic Res Cardiol 2023;118:49.
57. Owesny P, Grune T. The link between obesity and aging - insights into cardiac energy metabolism. Mech Ageing Dev 2023;216:111870.
58. Boardman NT, Trani G, Scalabrin M, Romanello V, Wüst RCI. Intracellular to interorgan mitochondrial communication in striated muscle in health and disease. Endocr Rev 2023;44:668-92.
59. Fernando R, Shindyapina AV, Ost M, et al. Downregulation of mitochondrial metabolism is a driver for fast skeletal muscle loss during mouse aging. Commun Biol 2023;6:1240.
60. Santulli G, Monaco G, Parra V, Morciano G. Editorial: mitochondrial remodeling and dynamic inter-organellar contacts in cardiovascular physiopathology. Front Cell Dev Biol 2021;9:679725.
61. Parra V, Monaco G, Morciano G, Santulli G. Editorial: mitochondrial remodeling and dynamic inter-organellar contacts in cardiovascular physiopathology-volume II. Front Cell Dev Biol 2023;11:1240207.
62. Katti P, Love-Rutledge S, Murray SA, Hinton A Jr. Editorial: the role of mitochondrial endoplasmic reticulum contact sites in human health and disease. Front Mol Biosci 2023;10:1223354.
63. Santulli G, Lewis D, des Georges A, Marks AR, Frank J. Ryanodine receptor structure and function in health and disease. Subcell Biochem 2018;87:329-52.
64. Fernandez-Sanz C, Ruiz-Meana M, Miro-Casas E, et al. Defective sarcoplasmic reticulum-mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis 2014;5:e1573.
66. Galizzi G, Di Carlo M. Insulin and its key role for mitochondrial function/dysfunction and quality control: a shared link between dysmetabolism and neurodegeneration. Biology 2022;11:943.
67. Cheng H, Gang X, He G, et al. The molecular mechanisms underlying mitochondria-associated endoplasmic reticulum membrane-induced insulin resistance. Front Endocrinol 2020;11:592129.
68. Peart JN, Pepe S, Reichelt ME, et al. Dysfunctional survival-signaling and stress-intolerance in aged murine and human myocardium. Exp Gerontol 2014;50:72-81.
69. Mendoza A, Karch J. Keeping the beat against time: mitochondrial fitness in the aging heart. Front Aging 2022;3:951417.
70. Paolocci N, Biondi R, Bettini M, et al. Oxygen radical-mediated reduction in basal and agonist-evoked NO release in isolated rat heart. J Mol Cell Cardiol 2001;33:671-9.
72. Jang S, Javadov S. Elucidating the contribution of ETC complexes I and II to the respirasome formation in cardiac mitochondria. Sci Rep 2018;8:17732.
73. Robb EL, Hall AR, Prime TA, et al. Control of mitochondrial superoxide production by reverse electron transport at complex I. J Biol Chem 2018;293:9869-79.
74. Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial electron transport chain, ROS generation and uncoupling (review). Int J Mol Med 2019;44:3-15.
75. Kausar S, Wang F, Cui H. The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cells 2018;7:274.
76. Chaudhary P, Janmeda P, Docea AO, et al. Oxidative stress, free radicals and antioxidants: potential crosstalk in the pathophysiology of human diseases. Front Chem 2023;11:1158198.
77. Jena AB, Samal RR, Bhol NK, Duttaroy AK. Cellular red-ox system in health and disease: the latest update. Biomed Pharmacother 2023;162:114606.
78. Hasanuzzaman M, Bhuyan MHMB, Zulfiqar F, et al. Reactive oxygen species and antioxidant defense in plants under abiotic stress: revisiting the crucial role of a universal defense regulator. Antioxidants 2020;9:681.
79. Gauthier LD, Greenstein JL, O’Rourke B, Winslow RL. An integrated mitochondrial ROS production and scavenging model: implications for heart failure. Biophys J 2013;105:2832-42.
80. Jomova K, Raptova R, Alomar SY, et al. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: chronic diseases and aging. Arch Toxicol 2023;97:2499-574.
81. Sharifi-Rad M, Anil Kumar NV, Zucca P, et al. Lifestyle, oxidative stress, and antioxidants: back and forth in the pathophysiology of chronic diseases. Front Physiol 2020;11:694.
82. Snezhkina AV, Kudryavtseva AV, Kardymon OL, et al. ROS generation and antioxidant defense systems in normal and malignant cells. Oxid Med Cell Longev 2019;2019:6175804.
83. Wang H, Liu C, Zhao Y, Gao G. Mitochondria regulation in ferroptosis. Eur J Cell Biol 2020;99:151058.
84. Rizvi F, Preston CC, Emelyanova L, et al. Effects of aging on cardiac oxidative stress and transcriptional changes in pathways of reactive oxygen species generation and clearance. J Am Heart Assoc 2021;10:e019948.
85. Napolitano G, Fasciolo G, Venditti P. Mitochondrial management of reactive oxygen species. Antioxidants 2021;10:1824.
86. Nulton-Persson AC, Szweda LI. Modulation of mitochondrial function by hydrogen peroxide. J Biol Chem 2001;276:23357-61.
87. Zorov DB, Popkov VA, Zorova LD, et al. Mitochondrial aging: is there a mitochondrial clock? J Gerontol A Biol Sci Med Sci 2017;72:1171-9.
88. Silachev DN, Plotnikov EY, Pevzner IB, et al. The mitochondrion as a key regulator of ischaemic tolerance and injury. Heart Lung Circ 2014;23:897-904.
89. Stanley BA, Sivakumaran V, Shi S, et al. Thioredoxin reductase-2 is essential for keeping low levels of H2O2 emission from isolated heart mitochondria. J Biol Chem 2011;286:33669-77.
90. Vue Z, Garza-Lopez E, Neikirk K, et al. 3D reconstruction of murine mitochondria reveals changes in structure during aging linked to the MICOS complex. Aging Cell 2023;22:e14009.
91. Warraich UE, Hussain F, Kayani HUR. Aging - oxidative stress, antioxidants and computational modeling. Heliyon 2020;6:e04107.
92. Checa J, Aran JM. Reactive Oxygen species: drivers of physiological and pathological processes. J Inflamm Res 2020;13:1057-73.
93. Juan CA, Pérez de la Lastra JM, Plou FJ, Pérez-Lebeña E. The chemistry of reactive oxygen species (ROS) revisited: outlining their role in biological macromolecules (DNA, Lipids and Proteins) and induced pathologies. Int J Mol Sci 2021;22:4642.
94. Anderson R, Richardson GD, Passos JF. Mechanisms driving the ageing heart. Exp Gerontol 2018;109:5-15.
95. Toba H, Lindsey ML. Extracellular matrix roles in cardiorenal fibrosis: potential therapeutic targets for CVD and CKD in the elderly. Pharmacol Ther 2019;193:99-120.
96. Umanskaya A, Santulli G, Xie W, Andersson DC, Reiken SR, Marks AR. Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc Natl Acad Sci USA 2014;111:15250-5.
97. Santulli G, Xie W, Reiken SR, Marks AR. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci USA 2015;112:11389-94.
98. Schriner SE, Linford NJ, Martin GM, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005;308:1909-11.
99. Mao P, Manczak M, Calkins MJ, et al. Mitochondria-targeted catalase reduces abnormal APP processing, amyloid β production and BACE1 in a mouse model of Alzheimer’s disease: implications for neuroprotection and lifespan extension. Hum Mol Genet 2012;21:2973-90.
100. Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal 2010;12:503-35.
101. Cardoso AC, Lam NT, Savla JJ, et al. Mitochondrial substrate utilization regulates cardiomyocyte cell cycle progression. Nat Metab 2020;2:167-78.
102. Linnane AW, Marzuki S, Ozawa T, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet 1989;1:642-5.
103. Druzhyna NM, Wilson GL, LeDoux SP. Mitochondrial DNA repair in aging and disease. Mech Ageing Dev 2008;129:383-90.
104. Gottlieb RA, Gustafsson AB. Mitochondrial turnover in the heart. Biochim Biophys Acta 2011;1813:1295-301.
105. Cortopassi GA, Arnheim N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res 1990;18:6927-33.
106. Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J 2005;19:419-21.
107. Ungvari Z, Orosz Z, Labinskyy N, et al. Increased mitochondrial H2O2 production promotes endothelial NF-kappaB activation in aged rat arteries. Am J Physiol Heart Circ Physiol 2007;293:H37-47.
108. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol 2018;15:505-22.
109. Ungvari Z, Kaley G, de Cabo R, Sonntag WE, Csiszar A. Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci 2010;65:1028-41.
110. Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A. Mechanisms of vascular aging. Circ Res 2018;123:849-67.
111. Donato AJ, Black AD, Jablonski KL, Gano LB, Seals DR. Aging is associated with greater nuclear NF kappa B, reduced I kappa B alpha, and increased expression of proinflammatory cytokines in vascular endothelial cells of healthy humans. Aging Cell 2008;7:805-12.
112. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 2015;21:677-87.
113. Bursch W, Ellinger A, Gerner C, Frohwein U, Schulte-Hermann, R. Programmed cell death (PCD). Apoptosis, autophagic PCD, or others? Ann N Y Acad Sci 2000;926:1-12.
114. Orogo AM, Gustafsson ÅB. Therapeutic targeting of autophagy: potential and concerns in treating cardiovascular disease. Circ Res 2015;116:489-503.
115. Xiong J, Luu TTT, Venkatachalam K, Du G, Zhu MX. Glutamine produces ammonium to tune lysosomal pH and regulate lysosomal function. Cells 2022;12:80.
116. Yin Z, Liu X, Ariosa A, et al. Psp2, a novel regulator of autophagy that promotes autophagy-related protein translation. Cell Res 2019;29:994-1008.
117. Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 2012;393:547-64.
119. Terman A, Brunk UT. Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc Res 2005;68:355-65.
120. Sneve ML, Overbye A, Fengsrud M, Seglen PO. Comigration of two autophagosome-associated dehydrogenases on two-dimensional polyacrylamide gels. Autophagy 2005;1:157-62.
122. Majeski AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol 2004;36:2435-44.
123. Santulli G. Cardioprotective effects of autophagy: eat your heart out, heart failure! Sci Transl Med 2018;10:eaau0462.
124. Bielawska M, Warszyńska M, Stefańska M, Błyszczuk P. Autophagy in heart failure: insights into mechanisms and therapeutic implications. J Cardiovasc Dev Dis 2023;10:352.
125. Santovito D, Steffens S, Barachini S, Madonna R. Autophagy, innate immunity, and cardiac disease. Front Cell Dev Biol 2023;11:1149409.
127. Sciarretta S, Boppana VS, Umapathi M, Frati G, Sadoshima J. Boosting autophagy in the diabetic heart: a translational perspective. Cardiovasc Diagn Ther 2015;5:394-402.
128. Kaludercic N, Maiuri MC, Kaushik S, et al. Comprehensive autophagy evaluation in cardiac disease models. Cardiovasc Res 2020;116:483-504.
129. Wang H, Wang L, Hu F, et al. Neuregulin-4 attenuates diabetic cardiomyopathy by regulating autophagy via the AMPK/mTOR signalling pathway. Cardiovasc Diabetol 2022;21:205.
130. Pyo JO, Yoo SM, Ahn HH, et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun 2013;4:2300.
131. Bhuiyan MS, Pattison JS, Osinska H, et al. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest 2013;123:5284-97.
133. Hariharan N, Ikeda Y, Hong C, et al. Autophagy plays an essential role in mediating regression of hypertrophy during unloading of the heart. PLoS One 2013;8:e51632.
134. Cao DJ, Jiang N, Blagg A, et al. Mechanical unloading activates FoxO3 to trigger Bnip3-dependent cardiomyocyte atrophy. J Am Heart Assoc 2013;2:e000016.
135. Yang YP, Liang ZQ, Gu ZL, Qin ZH. Molecular mechanism and regulation of autophagy. Acta Pharmacol Sin 2005;26:1421-34.
136. Yuan Q, Chen Z, Santulli G, et al. Functional role of Calstabin2 in age-related cardiac alterations. Sci Rep 2014;4:7425.
137. Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest 2015;125:25-32.
138. Dossou AS, Basu A. The Emerging Roles of mTORC1 in macromanaging autophagy. Cancers 2019;11:1422.
139. Deleyto-Seldas N, Efeyan A. The mTOR-autophagy axis and the control of metabolism. Front Cell Dev Biol 2021;9:655731.
140. Abdellatif M, Trummer-Herbst V, Heberle AM, et al. Fine-tuning cardiac insulin-like growth factor 1 receptor signaling to promote health and longevity. Circulation 2022;145:1853-66.
142. Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010;141:290-303.
143. Santulli G, Totary-Jain H. Tailoring mTOR-based therapy: molecular evidence and clinical challenges. Pharmacogenomics 2013;14:1517-26.
144. Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012;32:2-11.
145. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 2011;13:1016-23.
146. Jung KW, Won YJ, Kong HJ, Oh CM, Seo HG, Lee JS. Prediction of cancer incidence and mortality in Korea, 2013. Cancer Res Treat 2013;45:15-21.
147. Füllgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol 2014;15:65-74.
148. Zhang Y, Wang X, Li XK, et al. Sirtuin 2 deficiency aggravates ageing-induced vascular remodelling in humans and mice. Eur Heart J 2023;44:2746-59.
149. Packer M. Cardioprotective effects of Sirtuin-1 and its downstream effectors: potential role in mediating the heart failure benefits of SGLT2 (sodium-glucose cotransporter 2) inhibitors. Circ Heart Fail 2020;13:e007197.
150. Alcendor RR, Gao S, Zhai P, et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res 2007;100:1512-21.
151. Peng J, Xiong J, Cui C, et al. Maternal eicosapentaenoic acid feeding decreases placental lipid deposition and improves the homeostasis of oxidative stress through a Sirtuin-1 (SIRT1) independent manner. Mol Nutr Food Res 2019;63:e1900343.
152. Karbasforooshan H, Karimi G. The role of SIRT1 in diabetic cardiomyopathy. Biomed Pharmacother 2017;90:386-92.
153. Hsu CP, Zhai P, Yamamoto T, et al. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation 2010;122:2170-82.
154. Wang B, Yang Q, Sun YY, et al. Resveratrol-enhanced autophagic flux ameliorates myocardial oxidative stress injury in diabetic mice. J Cell Mol Med 2014;18:1599-611.
155. Bennett CF, Kaeberlein M. The mitochondrial unfolded protein response and increased longevity: cause, consequence, or correlation? Exp Gerontol 2014;56:142-6.
156. Weng H, Ma Y, Chen L, et al. A new vision of mitochondrial unfolded protein response to the sirtuin family. Curr Neuropharmacol 2020;18:613-23.
157. Ji T, Zhang X, Xin Z, et al. Does perturbation in the mitochondrial protein folding pave the way for neurodegeneration diseases? Ageing Res Rev 2020;57:100997.
158. Cilleros-Holgado P, Gómez-Fernández D, Piñero-Pérez R, et al. Mitochondrial quality control via mitochondrial unfolded protein response (mtUPR) in ageing and neurodegenerative diseases. Biomolecules 2023;13:1789.
159. Chen Q, Li L, Samidurai A, et al. Acute endoplasmic reticulum stress-induced mitochondria respiratory chain damage: the role of activated calpains. FASEB J 2024;38:e23404.
160. Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J 2021;40:e104705.
161. Siasos G, Tsigkou V, Kosmopoulos M, et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann Transl Med 2018;6:256.
162. Peoples JN, Saraf A, Ghazal N, Pham TT, Kwong JQ. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med 2019;51:1-13.
163. Bhullar SK, Dhalla NS. Status of mitochondrial oxidative phosphorylation during the development of heart failure. Antioxidants 2023;12:1941.
164. Chistiakov DA, Shkurat TP, Melnichenko AA, Grechko AV, Orekhov AN. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann Med 2018;50:121-7.
165. Scheibye-knudsen M, Fang EF, Croteau DL, Wilson DM, Bohr VA. Protecting the mitochondrial powerhouse. Trends Cell Biology 2015;25:158-70.
166. Stamerra CA, Di Giosia P, Giorgini P, Ferri C, Sukhorukov VN, Sahebkar A. Mitochondrial dysfunction and cardiovascular disease: pathophysiology and emerging therapies. Oxid Med Cell Longev 2022;2022:9530007.
167. Kubli DA, Gustafsson ÅB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res 2012;111:1208-21.
168. Liang W, Moyzis AG, Lampert MA, Diao RY, Najor RH, Gustafsson ÅB. Aging is associated with a decline in Atg9b-mediated autophagosome formation and appearance of enlarged mitochondria in the heart. Aging Cell 2020;19:e13187.
169. Wei X, Wu YE, Wang W, Zhang S, Liu D, Liu H. Decreased dynamin-related protein 1-related mitophagy induces myocardial apoptosis in the aging heart. Acta Biochim Biophys Sin 2021;53:1354-66.
170. Fernández-Ortiz M, Sayed RKA, Fernández-Martínez J, et al. Melatonin/Nrf2/NLRP3 connection in mouse heart mitochondria during aging. Antioxidants 2020;9:1187.
171. Lee Y, Lee HY, Hanna RA, Gustafsson ÅB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol 2011;301:H1924-31.
172. Hamacher-Brady A, Brady NR, Logue SE, et al. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ 2007;14:146-57.
174. Wang Y, Xu Y, Guo W, et al. Ablation of Shank3 alleviates cardiac dysfunction in aging mice by promoting CaMKII activation and Parkin-mediated mitophagy. Redox Biol 2022;58:102537.
175. Shi R, Guberman M, Kirshenbaum LA. Mitochondrial quality control: the role of mitophagy in aging. Trends Cardiovasc Med 2018;28:246-60.
176. Jankauskas S, Kansakar U, De Donato A, Mone P, Varzideh F, Santulli G. Parkin controls cardiac function in obesity by regulating mitochondrial calcium uptake. JACC Basic Transl Sci 2022;7:797-9.
177. Zhu Q, Combs ME, Liu J, et al. GRAF1 integrates PINK1-Parkin signaling and actin dynamics to mediate cardiac mitochondrial homeostasis. Nat Commun 2023;14:8187.
178. Soh JEC, Shimizu A, Molla MR, et al. RhoA rescues cardiac senescence by regulating Parkin-mediated mitophagy. J Biol Chem 2023;299:102993.
179. LaRocca TJ, Hearon CM Jr, Henson GD, Seals DR. Mitochondrial quality control and age-associated arterial stiffening. Exp Gerontol 2014;58:78-82.
180. Kubli DA, Quinsay MN, Gustafsson AB. Parkin deficiency results in accumulation of abnormal mitochondria in aging myocytes. Commun Integr Biol 2013;6:e24511.
181. Huang W, Xie W, Zhong H, et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes acute liver injury induced by heat stroke. Front Immunol 2022;13:859231.
182. Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci USA 2011;108:9572-7.
183. Poznyak AV, Kirichenko TV, Borisov EE, Shakhpazyan NK, Kartuesov AG, Orekhov AN. Mitochondrial implications in cardiovascular aging and diseases: the specific role of mitochondrial dynamics and shifts. Int J Mol Sci 2022;23:2951.
184. Lu X, Gong Y, Hu W, et al. Ultrastructural and proteomic profiling of mitochondria-associated endoplasmic reticulum membranes reveal aging signatures in striated muscle. Cell Death Dis 2022;13:296.
185. Lozhkin A, Vendrov AE, Ramos-Mondragón R, et al. Mitochondrial oxidative stress contributes to diastolic dysfunction through impaired mitochondrial dynamics. Redox Biol 2022;57:102474.
186. Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci 2001;114:867-74.
187. Di Nottia M, Verrigni D, Torraco A, Rizza T, Bertini E, Carrozzo R. Mitochondrial dynamics: molecular mechanisms, related primary mitochondrial disorders and therapeutic approaches. Genes 2021;12:247.
188. Zacharioudakis E, Biris N, Garner TP, et al. Direct small molecule activation of mitofusins. bioRxiv Forthcoming 2018.
189. Zerihun M, Sukumaran S, Qvit N. The Drp1-mediated mitochondrial fission protein interactome as an emerging core player in mitochondrial dynamics and cardiovascular disease therapy. Int J Mol Sci 2023;24:5785.
190. Ihenacho UK, Meacham KA, Harwig MC, Widlansky ME, Hill RB. Mitochondrial fission protein 1: emerging roles in organellar form and function in health and disease. Front Endocrinol 2021;12:660095.
191. Hu Q, Zhang H, Gutiérrez Cortés N, et al. Increased Drp1 acetylation by lipid overload induces cardiomyocyte death and heart dysfunction. Circ Res 2020;126:456-70.
192. Basu K, Lajoie D, Aumentado-Armstrong T, et al. Molecular mechanism of DRP1 assembly studied in vitro by cryo-electron microscopy. PLoS One 2017;12:e0179397.
193. Ljubicic V, Menzies KJ, Hood DA. Mitochondrial dysfunction is associated with a pro-apoptotic cellular environment in senescent cardiac muscle. Mech Ageing Dev 2010;131:79-88.
194. Breitzig MT, Alleyn MD, Lockey RF, Kolliputi N. A mitochondrial delicacy: dynamin-related protein 1 and mitochondrial dynamics. Am J Physiol Cell Physiol 2018;315:C80-90.
195. Piquereau J, Caffin F, Novotova M, et al. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc Res 2012;94:408-17.
196. Cahill TJ, Leo V, Kelly M, et al. Resistance of dynamin-related protein 1 oligomers to disassembly impairs mitophagy, resulting in myocardial inflammation and heart failure. J Biol Chem 2015;290:25907-19.
197. Li Z, Zhang Z, Ren Y, et al. Aging and age-related diseases: from mechanisms to therapeutic strategies. Biogerontology 2021;22:165-87.
198. Zorov DB, Isaev NK, Plotnikov EY, et al. Perspectives of mitochondrial medicine. Biochemistry 2013;78:979-90.
199. Ramaccini D, Montoya-Uribe V, Aan FJ, et al. Mitochondrial function and dysfunction in dilated cardiomyopathy. Front Cell Dev Biol 2020;8:624216.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Mone P, Agyapong ED, Morciano G, Jankauskas SS, De Luca A, Varzideh F, Pinton P, Santulli G. Dysfunctional mitochondria elicit bioenergetic decline in the aged heart. J Cardiovasc Aging 2024;4:13. http://dx.doi.org/10.20517/jca.2023.50
AMA Style
Mone P, Agyapong ED, Morciano G, Jankauskas SS, De Luca A, Varzideh F, Pinton P, Santulli G. Dysfunctional mitochondria elicit bioenergetic decline in the aged heart. The Journal of Cardiovascular Aging. 2024; 4(2): 13. http://dx.doi.org/10.20517/jca.2023.50
Chicago/Turabian Style
Mone, Pasquale, Esther Densu Agyapong, Giampaolo Morciano, Stanislovas S. Jankauskas, Antonio De Luca, Fahimeh Varzideh, Paolo Pinton, Gaetano Santulli. 2024. "Dysfunctional mitochondria elicit bioenergetic decline in the aged heart" The Journal of Cardiovascular Aging. 4, no.2: 13. http://dx.doi.org/10.20517/jca.2023.50
ACS Style
Mone, P.; Agyapong ED.; Morciano G.; Jankauskas SS.; De Luca A.; Varzideh F.; Pinton P.; Santulli G. Dysfunctional mitochondria elicit bioenergetic decline in the aged heart. J. Cardiovasc. Aging. 2024, 4, 13. http://dx.doi.org/10.20517/jca.2023.50
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 10 clicks
Cite This Article 10 clicks

 Like This Article 14
likes
Like This Article 14
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.