
Extracellular vesicles: the key to unlocking mechanisms of age-related vascular disease?
 0
0
Abstract
Aging is associated with the development of two ubiquitous, detrimental pathologies, vascular calcification and amyloidosis. These pathologies are characterized by the accumulation of toxic aggregates in the vessel extracellular matrix (ECM) in the form of crystalline calcium and phosphate mineral or insoluble protein fibrils, respectively. These aggregates impact ECM integrity, drive vascular stiffening, and can also cause cell death and phenotypic change in the cells that interact with them. The deposition of both calcification and amyloid requires a nucleus that can mediate the mineralization of calcium and phosphate, or amyloid aggregation from precursor proteins or peptides. Emerging evidence suggests that changes in the composition of the ECM associated with cellular senescence, as well as extracellular vesicle (EV) release, cargo-loading, trapping, and aggregation within the ECM, are common and synergistic mechanisms that regulate the development of these pathologies. Importantly, vascular smooth muscle cells (VSMCs) orchestrate the formation of both pathologies that commonly co-occur in the aging vasculature. Here, we outline the commonalities and differences in what is known about the genesis of calcification and amyloid, and highlight key questions and areas that remain unknown and require further investigation. The complex relationship between senescence, EVs, and the ECM, mediated by VSMCs, which drives the accumulation of HA and amyloid, could be a target for therapeutic intervention.
Keywords
INTRODUCTION
Advancing age is the major risk factor for cardiovascular disease (CVD). With age, the occurrence and prevalence of coronary heart disease, hypertension, and atherosclerosis increase dramatically, making CVD the leading cause of death worldwide, representing 30% of all deaths[1]. By 2050, the incidence of CVD could double in conjunction with increases in life expectancy and damaging trends, such as obesity and smoking[2]. A significant feature of vascular aging is increased vascular stiffness, which can worsen chronic conditions, such as hypertension, contributing to CVD-related mortality rates[3,4]. Pulse wave velocity (PWV), a clinical measure of arterial stiffness, is an independent indicator of cardiovascular risk and is used as a marker of vascular aging[5]. Age-related diseases, such as chronic kidney disease (CKD)[6], atherosclerosis[7], Alzheimer’s disease (AD)[8], and dementia[9], are all associated with increased PWV, which is a predictor of all-cause mortality as well as cardiovascular mortality[10,11], highlighting the significance of vascular stiffening in age-related decline.
Vascular calcification and vascular amyloidosis are two age-related pathologies that are highly prevalent in patients over the age of 60 years[12-14]. These pathologies affect both the intimal and medial layers of the vessel wall and accelerate vascular stiffening, thus promoting significant negative outcomes[15,16]. Both pathologies are mediated by vascular smooth muscle cells (VSMCs) and emerging data suggest that similar mechanisms may contribute to their progression with age. However, despite this dramatic association with age, their co-occurrence and possible common and/or interacting mechanisms have not been systematically investigated.
Vascular calcification is characterized by the accumulation of hydroxyapatite (HA), a calcium phosphate mineral [Ca3(PO4)2]. There is a strong association between age and medial calcification[17], with vascular calcification also being closely correlated to biological aging, as younger patients with calcification have advanced aging of the vasculature and the survival risk of a person significantly older[18]. Previously considered a passive, degenerative process associated with aging, it is now recognized as an active and dynamic process orchestrated by VSMCs and involving the upregulation of genes associated with both osteogenesis and chondrogenesis, with many similarities to the formation of bone[19].
Calcification occurs at two anatomical sites in the vessel wall, the intima and the media. Intimal calcification is linked with atherosclerosis and is characterized by HA accumulation with lipids, macrophages, and VSMCs. This causes inflammation, growth, and potentially destabilization of atherosclerotic plaques
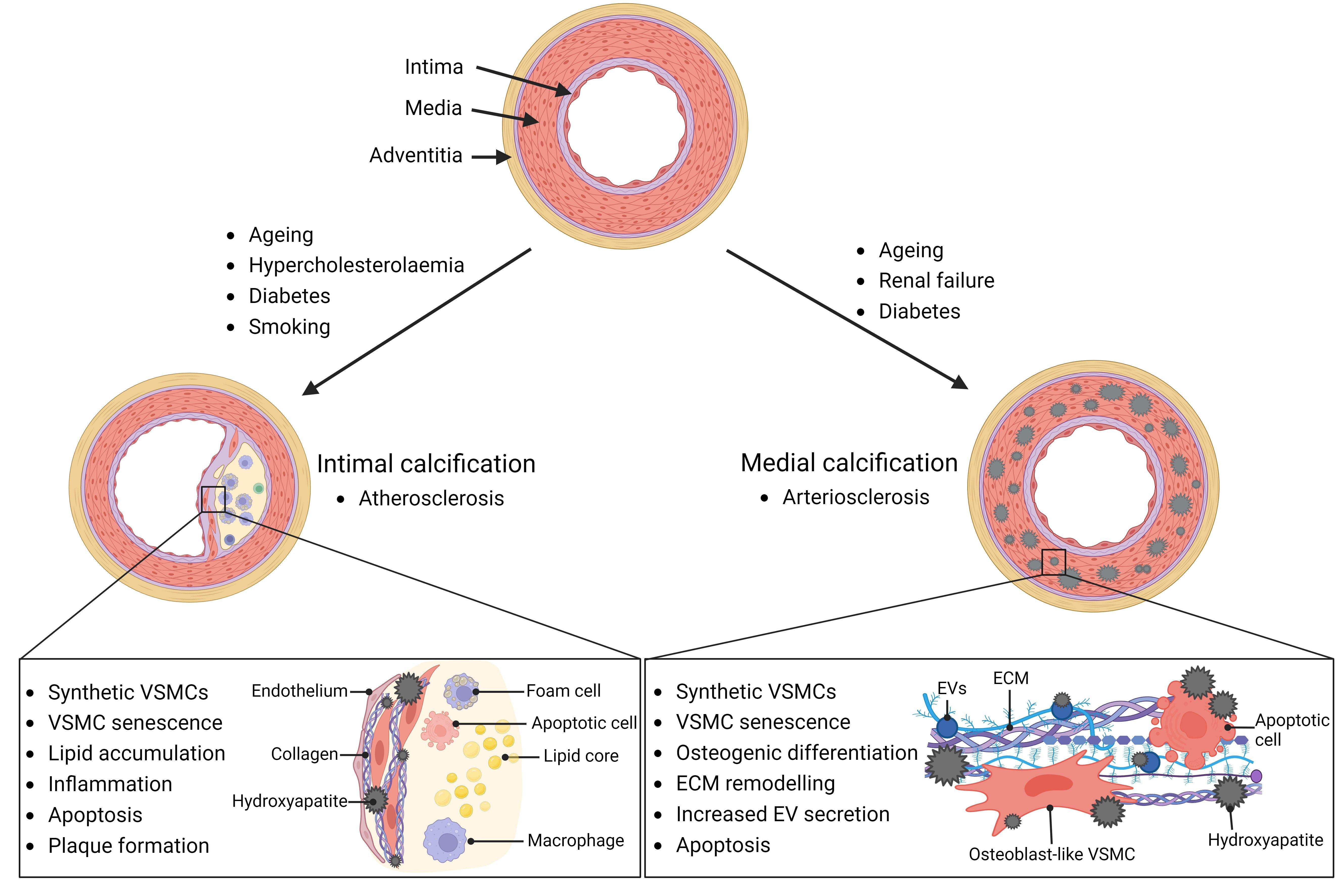
Figure 1. The vessel wall is organized into 3 layers. The intimal layer is made up of one layer of endothelial cells and the internal elastic lamina. The medial layer of a healthy vessel consists of contractile vascular smooth muscle cells (VSMCs), the extracellular matrix (ECM), and elastic fibers. The adventitia is the outer layer of connective tissue. Vascular calcification can occur at the intimal and medial layers with different risk factors and outcomes. Intimal calcification is commonly associated with atherosclerosis and is characterized by formation of a plaque with a fibrous cap and accumulation of lipids and inflammatory cells. Medial calcification often results in arteriosclerosis and vascular stiffening. VSMCs are the main mediators of medial calcification and can undergo phenotypic differentiation into a more synthetic, osteoblast-like phenotype, characterized by extracellular matrix (ECM) remodeling and extracellular vesicle (EV) secretion. (Created with BioRender.com)
Amyloidosis is the accumulation of insoluble, fibrous amyloid, formed from the aggregation of soluble proteins or peptide monomers into β-sheets[24]. Vascular amyloidosis occurs in the vessel intima and media and several amyloids have been described in the cardiovascular system[25]. Cardiovascular amyloids are commonly classified as age-related and are often not associated with antecedent disease, with age being the only risk factor[26,27]. In the vessel wall, the most common amyloids are aortic medial amyloid (AMA), amyloid transthyretin (ATTR), apolipoprotein A-1, and amyloid light chain (LC)[28,29]. To date, there has been limited research investigating the pathological role of amyloid deposits in the vasculature. The two amyloids that have received the most attention are medin, the cause of AMA, which is present in the aorta of over 97% of Caucasian people over 50 years old[30], and Aβ, the same amyloid that causes Alzheimer’s Disease (AD), that accumulates in leptomeningeal vessels of the brain in cerebral amyloid angiopathy (CAA). Previous studies have suggested that medin accumulation in the vessel wall can reduce arterial elasticity through its interaction with elastic fibers[31], while stiffening of cerebral arteries is thought to accelerate Aβ accumulation in the brain[32,33]. However, more studies are required to delineate exactly the effects of amyloidosis in the vasculature.
A key feature of aging is the accumulation of senescent cells[34]. These cells show a number of specific features including exit from the cell cycle, morphology change, persistent DNA damage, and activation of the senescence-associated secretory phenotype (SASP), leading to the release of inflammatory mediators and proteases that can act remotely to accelerate cellular dysfunction and aging. In the vessel wall, senescent cells accumulate with age and premature senescence of VSMCs is a feature of young patients with calcified arteries, such as children on dialysis[18,35]. Extensive research has been conducted on the impact of senescence on vascular calcification[36]; however, there remains a lack of knowledge regarding the relationship between senescence, calcification, and amyloidosis, despite several forms of pathological amyloidosis being associated with aging.
Senescent VSMCs have also been shown to increase the secretion of extracellular vesicles (EVs)[37]. The role of EVs in calcification and amyloidosis in the vasculature is interesting as both pathologies require a nucleus to initiate either calcification or amyloid formation. VSMC-derived EVs and exosomes have been implicated in the deposition and nucleation of both HA and amyloid[38-40], but the precise mechanisms governing what initiates EVs to mineralize, aggregate protein, or both are unknown. However, it is clear that EVs are an important trigger for both pathologies and could account for their concomitant development in the vessel wall with age.
Building on these key facts, in this review, we will focus on the role of senescence and the involvement of EVs in driving these pathologies. The co-occurrence of calcification and amyloidosis, particularly in the vasculature, and their common and reciprocal mechanisms need further investigation to tease apart their roles in CVD and identify novel therapeutic targets to alleviate vascular stiffening.
MECHANISMS OF VASCULAR CALCIFICATION AND AMYLOID DEPOSITION: A KEY ROLE FOR VSMCS
Vascular calcification
VSMCs in the arterial tunica media have a contractile phenotype, providing dynamic adaptability to blood volume changes occurring in the vasculature. In healthy adults, VSMCs exhibit a contractile phenotype with low proliferation, but with aging and the occurrence of chronic inflammation and structural damage, VSMCs switch to a more proliferative/synthetic phenotype and migrate to the site of injury [Figure 2][41,42]. This synthetic characteristic provides cytokines and growth factors and induces overexpression of elastin, collagens, proteoglycans, and other ECM components to repair the damaged wall[43]. Synthetic VSMCs secrete EVs containing calcification inhibitors, β1 integrin, and ECM proteins such as fibronectin and collagen VI, which stimulate cell migration and invasion[44].
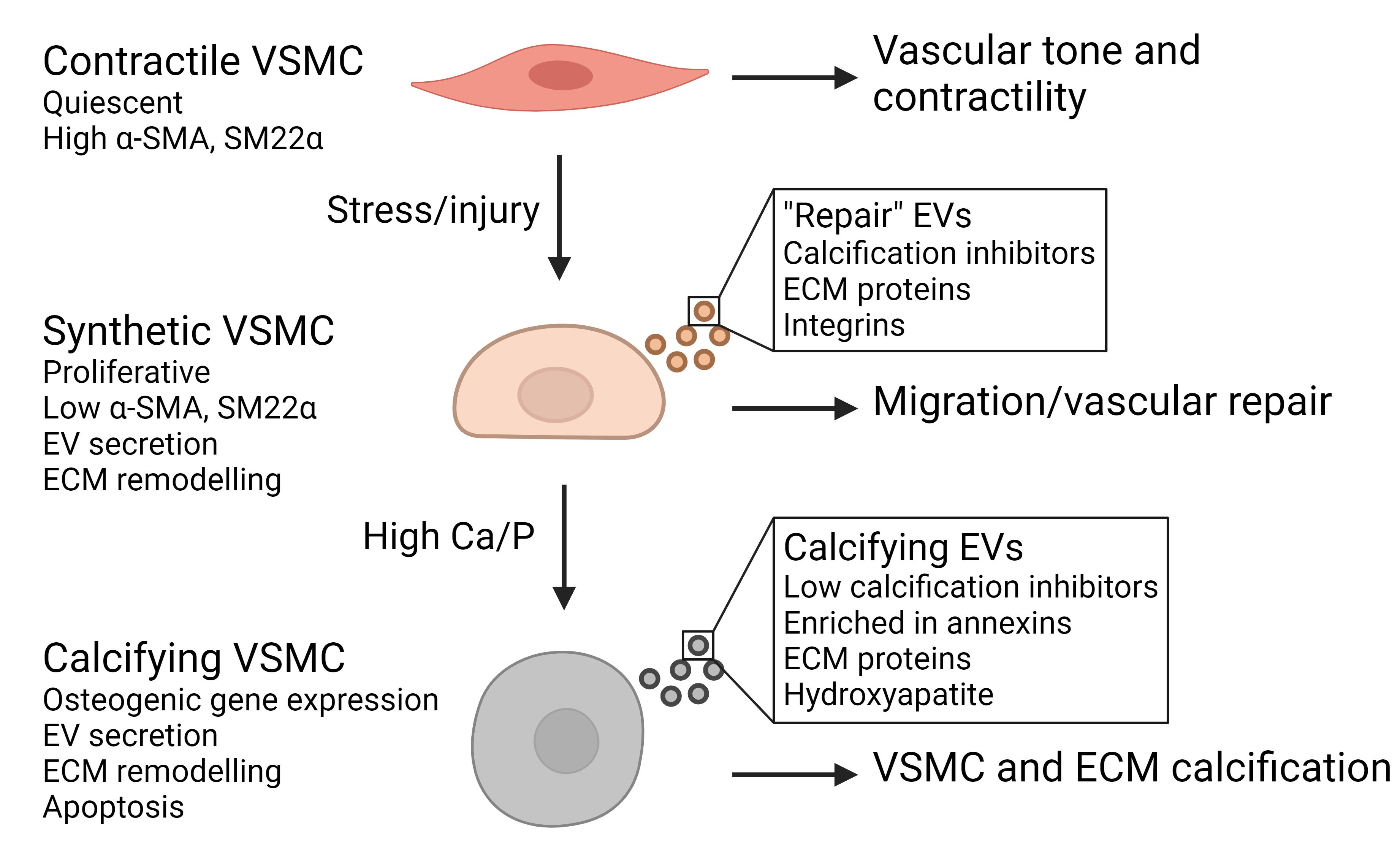
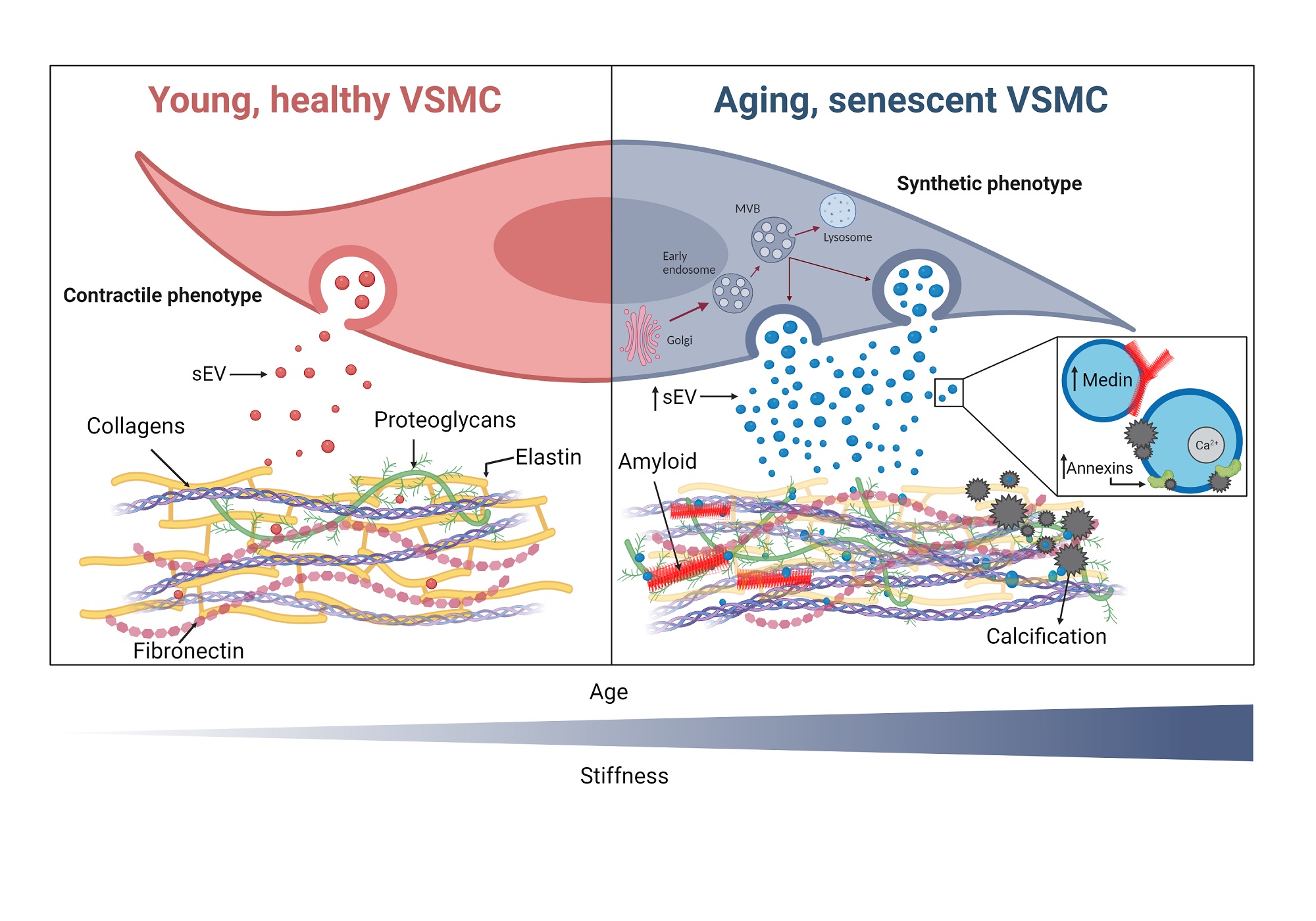
Figure 2. Contractile vascular smooth muscle cells (VSMCs) express high levels of contractility markers, including α-smooth muscle actin (α-SMA) and smooth muscle protein 22α (SM22α), to regulate vascular tone. Synthetic VSMCs have low contractility, proliferation and secrete extracellular vesicles (EVs) that can promote VSMC migration and invasion to drive vascular repair after injury. Elevated calcium and phosphate levels (Ca/P) can induce an osteogenic VSMC phenotype with bone-like markers, extracellular matrix (ECM) deposition, and apoptosis. Osteogenic VSMCs secrete calcifying EVs, which form a nidus for mineralization due to the loss of calcification inhibitors and being rich in annexins and phosphatidylserine.
Calcification is a dynamic process involving VSMC osteogenic differentiation, with overexpression of pro-osteogenic genes, enhancing the accumulation of HA [Figure 2][16,45]. During the initial stages of calcification, runt-related transcription factor (Runx2), which regulates osteogenic differentiation during the physiological formation of bones, is upregulated. In addition, other transcription factors (TF) and morphogens that can drive osteo/chondrogenic pathways, such as Sox9 or Msx2, which are involved in cartilage differentiation and bone maturation, respectively, and bone morphogenetic protein 2 (BMP2) are upregulated[46,47]. These TFs also regulate the expression of a number of proteins such as matrix metalloproteinases (MMPs), osteocalcin (OCN), collagen type 1-alpha1 (COL1α1), bone sialoprotein (BSP), and alkaline phosphatase (ALP) that can regulate mineral growth[48]. VSMC osteogenic differentiation is accompanied by downregulation of VSMC contractile genes, such as smooth muscle protein 22-α (SM22α) and α-smooth muscle actin (α-SMA), which also control cell motility and shape[49].
VSMCs can respond to pathological stimuli by expressing calcification inhibitors. For instance, matrix Gla
Vascular amyloid
Like HA, amyloid fibril formation is a nucleation-dependent mechanism. It can be initiated by soluble, cleaved peptide monomers or destabilized protein structures, which are susceptible to aggregation into oligomers, which may induce cell death, or further aggregation into amyloid. Partial unfolding of proteins can occur at high temperatures or low pH[52]. Aggregation of denatured monomers or amyloidogenic peptides occurs slowly during the lag phase and occurs when hydrophobic regions of proteins, which are usually embedded in the protein structure, become exposed through cleavage or denaturation and begin to aggregate with other hydrophobic regions or peptides [Figure 3]. The growth of unstructured aggregates and amyloid seeds into mature fibrils occurs exponentially during the growth phase to more thermodynamically stable fibrils[53]. Amyloid plaques generally comprise several components, the fibrils from denatured protein or peptide precursor, non-fibrillar serum amyloid P component (SAP), glycosaminoglycans (GAGs), heparin sulfate proteoglycans (HSPGs), and apolipoprotein E (apoE). Previous research suggests SAP and apoE stabilize amyloid fibrils and protect them from degradation by increasing fibril strength and density[54].
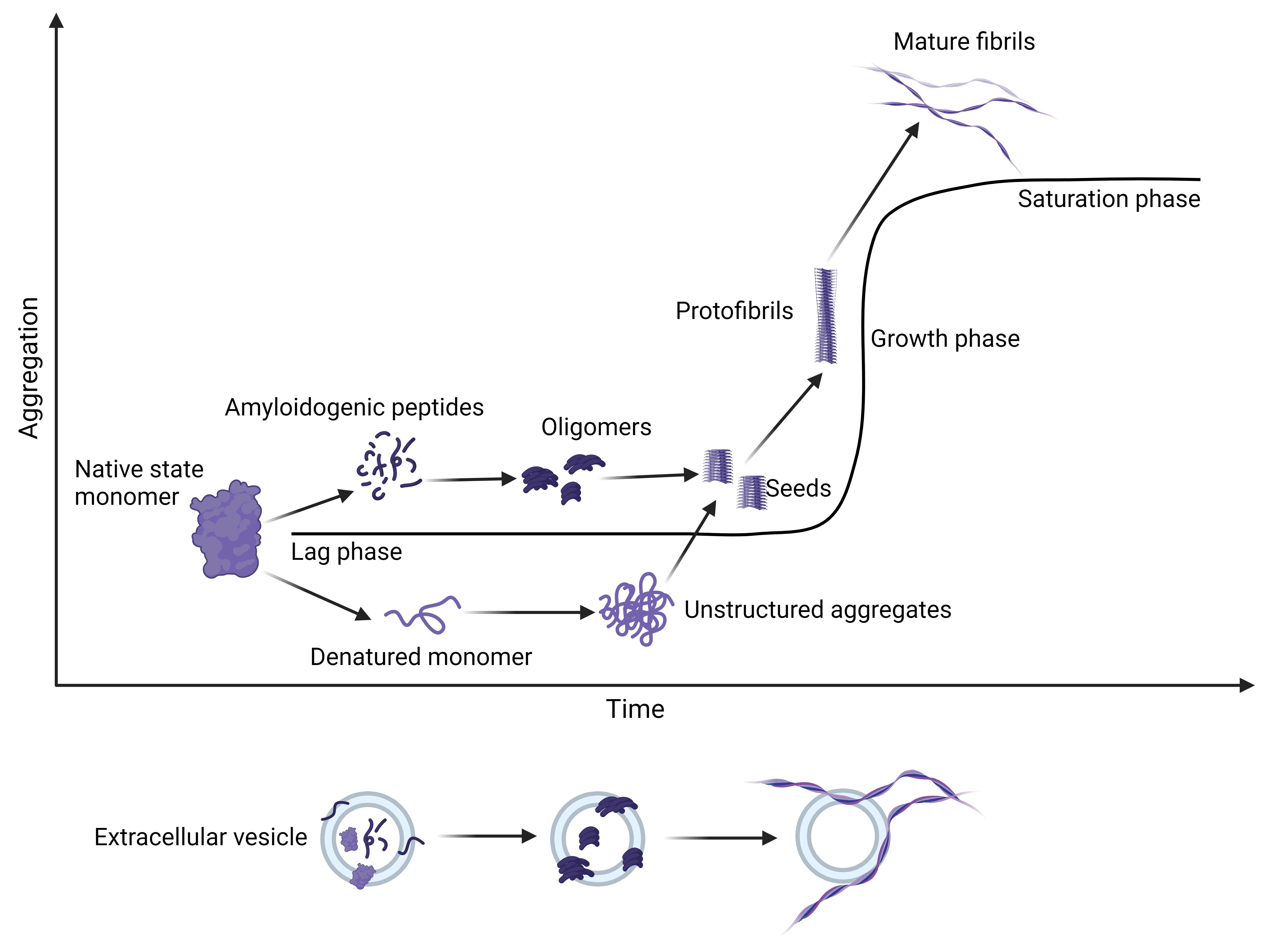
Figure 3. Amyloid fibrils are formed by denaturation of a native state protein to form a denatured monomer or cleavage to form amyloidogenic peptides. These aggregate slowly to form amyloid seeds during the lag phase. The amyloid seeds rapidly aggregate into β-sheets in the exponential growth phase to form insoluble amyloid fibrils. Extracellular vesicles have been identified as mediators of amyloid formation as they contain several different amyloid precursor proteins or peptides. They can accelerate the aggregation of amyloid peptides by acting as a nucleus for aggregation and fibril growth. (Created with BioRender.com)
AMA is a localized, aortic, age-related amyloid. It is a major cardiovascular amyloid, comprising a 50-amino acid peptide medin, produced by cleavage of milk fat globule epidermal growth factor (EGF) 8 (MFGE8)[30,31,55]. MFGE8 is highly expressed by VSMCs and accumulates in the vessel wall with age[56], with its accumulation also correlating with PWV and cardiovascular risk factors in diabetic patients[57]. It contains a domain with an RGD peptide that binds integrins to mediate several processes in VSMCs, including proliferation[58], osteogenic differentiation[59], and inflammation[60]. Medin is composed of a fragment from the C2-like domain of MFGE8, which can bind phospholipids. Amyloid deposits in the medial layer of the thoracic aorta are present in over 97% of Caucasian people over 50 years old[30]. Both MFGE8 and medin have been shown to bind to elastin, accounting for its accumulation in large arteries[61], and previous studies have suggested that medin accumulation in the vessel wall can reduce arterial elasticity through its interaction with elastic fibers[31].
Amyloid-β (Aβ) peptides are recognized as the primary driver of AD. Their accumulation leads to the formation of characteristic plaques within the aging brain parenchyma. The aggregation of Aβ peptides has also been identified in crucial cerebral blood vessels, such as the internal carotid artery, resulting in CAA[62]. Aβ accumulates predominantly in the medial and adventitial layers of leptomeningeal arteries, indicating a role of VSMCs in its deposition[63]. Cerebral arteries play a key role in the clearance of Aβ through the perivascular drainage pathway, which is reduced with age and in AD, leading to Aβ deposition in the ECM and its uptake by VSMCs, causing disruption of the vessel wall[64,65]. Furthermore, substantial amounts of Aβ can be detected in the circulation and accumulate in atherosclerotic plaques in the carotid artery and aorta[66,67]. Interestingly, circulating Aβ levels have been shown to be predictive of cardiovascular mortality and vascular disease progression in patients with coronary heart disease and were significantly associated with arterial stiffness and atherosclerosis[68,69]. However, despite its presence in the cardiovascular system and association with vascular disease, its pathological role and mechanisms of accumulation have not been investigated.
AGING AND SENESCENCE
Cellular senescence is a hallmark of vascular aging, calcification, and amyloidosis[34]. This physiological process is characterized by telomere shortening, DNA damage, and arrest of the cell cycle, with cells unable to respond correctly to mitotic stimuli. Cellular senescence triggers several cell cycle regulators, such as p53/p21 and p16/retinoblastoma protein tumor suppressor activation[70,71], and plays a crucial role in vascular aging through increased secretion of inflammatory cytokines, proteases, and growth factors, comprising the SASP.
Recent studies have demonstrated that VSMCs rapidly undergo osteogenic differentiation during aging[72,73], which could both be driven by, and contribute to, cellular senescence. VSMC senescence in vitro has been shown to enhance calcification and increase the expression of osteogenic genes, such as Runx2, ALP, and collagen 1[73]. In klotho knockout mice, a mouse model of aging, significantly upregulated expression of RUNX2 was shown in the calcified lesions in the aorta[74]. Interestingly, CKD patients have decreased klotho levels, which decline further as the disease progresses[75]. Senescent VSMCs and apoptotic bodies have also been detected in the medial layer of calcified arteries from children with CKD, with their vascular phenotype more closely resembling that of old subjects[45,76,77], highlighting the role of senescence in driving calcification during aging.
VSMC senescence also contributes to calcification by the release of inflammatory cytokines and growth factors. In vitro replicative senescence of VSMCs shows correlations with age-related morphological changes and DNA damage accumulation, which can lead to the overexpression of pro-osteogenic genes and downregulation of calcification inhibitors[72,73]. Inflammatory cytokines, particularly interleukins (ILs), are highly activated during the SASP, with IL-6 promoting calcification through osteogenic gene upregulation (Runx2, ALP, and MMP2) via extracellular related kinase (ERK) signaling pathways[78].
There is strong evidence showing the accumulation of senescent VSMCs in atherosclerotic plaques. VSMC senescence in atherosclerosis is thought to be induced by replication, resulting in telomere shortening, or stress-induced premature senescence (SIPS), caused by DNA damage and oxidative stress[79,80]. While it is not clear what role senescence-induced calcification has in plaque formation and stability, it has been shown to be closely correlated with cardiovascular events, providing further evidence for the importance of VSMC senescence in both the intima and media of the vessel wall[81,82].
Extracellular proteases are also components of the SASP. MMPs play an important role in both bone and vascular tissue remodeling[83]. Several studies have identified MMPs as promoters of vascular calcification[84,85]. MMPs degrade several key ECM components that are important in maintaining integrity of the vessel wall, such as elastin, fibronectin, and laminin. Previous research showed that the degradation of elastin preceded VSMC loss and vascular calcification in a model of CKD[86]. Therefore, enhanced matrix degradation, induced by SASP, could contribute to vascular calcification. In addition to matrix degradation, MMP2 and MMP9 have been shown to enhance vascular calcification through upregulation of BMP2[85].
To date, there is limited understanding of the contribution of senescence to amyloidosis, particularly in the cardiovascular system. Although AMA is an extremely prevalent and age-associated form of amyloidosis, the role of cellular aging in its development is under-researched. Accumulation of medin in the medial layer of human aortas has been strongly correlated with age[56], and a recent study showed VSMC senescence increased the production and secretion of medin in sEVs[37]. This resulted in increased accumulation of medin in the ECM, promoting its aggregation into amyloid fibrils, indicating a role of senescence in triggering AMA development.
Senescent cells have been detected in the vasculature in both AD patients and AD mouse models and are thought to contribute to increased permeability and loss of integrity of the blood-brain barrier[87]. VSMC senescence has not yet been investigated thoroughly in CAA, which overlaps significantly with AD. Cellular senescence plays a role in AD, although its precise mechanisms are not fully understood[88]. Various cell types, such as astrocytes, neurons, and endothelial cells, express senescence-associated proteins in AD brains, indicating a link between senescence and Aβ pathophysiology[89,90].
EXTRACELLULAR VESICLES
An important mechanism of vascular calcification is the secretion and deposition of EVs from VSMCs. EVs have also been linked to amyloid-related diseases in both physiological and pathological contexts, including AD[91,92] and AMA[37,93].
EVs are key mediators of intercellular communication, acting as vehicles for transporting proteins, lipids, and RNA[94]. Importantly, EVs comprise different subtypes of vesicles, often categorized by size, biogenesis, or isolation technique. Microvesicles (200-1,000 nm) outwardly bud off directly from the plasma membrane of cells, while small EVs (sEVs) or exosomes (10-200 nm) are formed within endosomes [Figure 4]. Apoptotic bodies (1-5 μm) are formed from cell fragments during apoptosis.
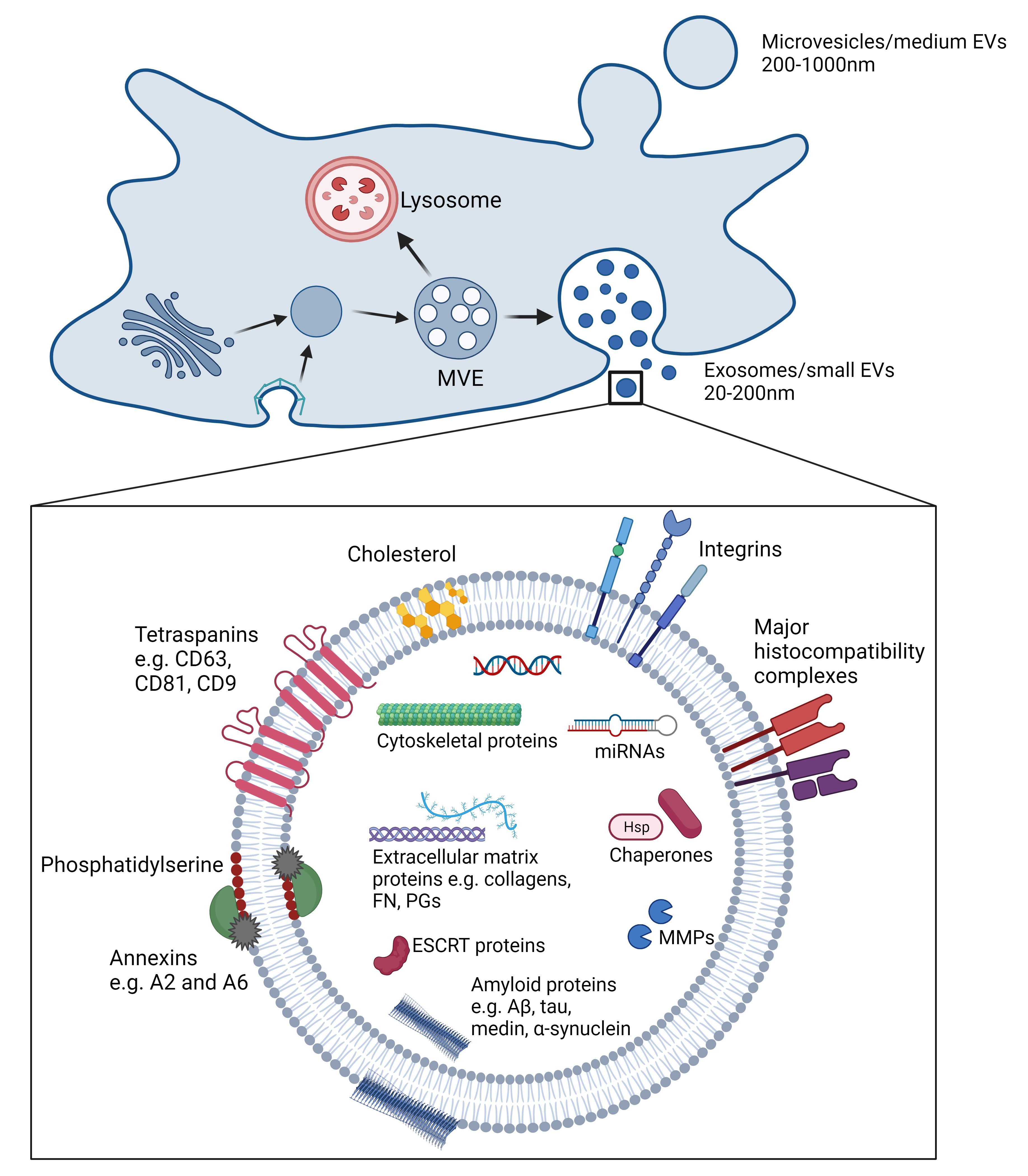
Figure 4. Blebbing of the plasma membrane causes the formation of microvesicles or medium extracellular vesicles (EVs). Exosomes or small EVs are formed in early endosomes from clathrin-coated vesicles or Golgi apparatus, where budding of the membrane to create intraluminal vesicles results in maturation into a multivesicular endosome (MVE). The MVE either fuses with lysosomes to be degraded or fuses with the plasma membrane to release the intraluminal vesicles as exosomes. Exosomes or small EVs contain a myriad of proteins and molecules, including microRNAs (miRNAs), extracellular matrix components, matrix metalloproteinases (MMPs), annexins, and several different amyloid proteins. (Created with BioRender.com)
Early endosomes are formed from inward budding of the cell membrane, forming clathrin-coated vesicles (CCVs), or from vesicles from the Golgi apparatus. Intraluminal vesicles (ILVs) are small, lipid bilayer-bound vesicles, which inwardly bud from the early endosomal membrane[95]. This becomes a multivesicular endosome (MVE) and the ILVs are released as exosomes, sometimes described as sEVs, when the MVEs fuse with the plasma membrane[96]. Alternatively, the ILVs are degraded by the fusion of the MVEs with lysosomes[96]. Exosomes are released from a variety of cell types, including blood cells, endothelial cells, and VSMCs and can interact with recipient cells through three different mechanisms: direct interaction between exosome transmembrane proteins and target cell receptors; fusion with the target plasma membrane, followed by the release of the contents into the cytosol; and internalization of exosomes into the cell[97]. Internalized exosomes can merge into endosomes, be released again into neighboring cells, or mature into lysosomes, leading to degradation[98].
Exosomes and sEVs consist of a lipid bilayer with several membrane proteins: major histocompatibility complexes (MHC), which are involved in antigen presentation; integrins and tetraspanins, for cell targeting and adhesion; annexins and flotillin, for membrane trafficking; and lipid rafts, which influence membrane fluidity and membrane protein and receptor trafficking[96]. They are loaded with a variety of proteins both on the surface and within the lumen, such as adhesion molecules and matrix-degrading enzymes, cytoskeletal proteins and enzymes, mRNA, and microRNAs.
Microvesicles are formed from direct budding of the plasma membrane and comprise a heterogeneous population of vesicles, differing based on cell type and function. The plasma membrane may undergo remodeling, meaning the composition of microvesicles can appear distinct from the parent cell[99]. Microvesicles can be packed with miRNAs, mRNAs, and proteins and, like exosomes or sEVs, can act as a mechanism for the transport of bioactive molecules and signaling.
Apoptotic bodies are formed during the disassembly and fragmentation of apoptotic cells. During apoptosis, cells shrink, and the intracellular contents become more densely packed. The plasma membrane then undergoes blebbing, forming apoptotic bodies, which contain components of the apoptotic cells, such as cytoplasm, organelles, chromatin, and other nuclear remnants[100]. The cargo of apoptotic bodies is very heterogeneous as contents are randomly packed into the vesicles.
EVs in vascular calcification
The change of VSMCs to a synthetic phenotype is a characteristic of vascular repair in calcifying conditions and aging and is associated with increased EV secretion[38]. Specifically, EV production and contents change with disease and stress, highlighting their roles in both normal, physiological, and pathological processes[101].
A previous study on vascular calcification demonstrated how calcium phosphate deposition in the medial layer of major arteries was mediated by exosomes. During HA deposition, there was a decreased concentration of calcification inhibitors and a disruption of vascular homeostasis[4]. Interestingly, VSMC-derived exosomes contain key calcification inhibitors, such as MGP and fetuin-A[102-104]. In chronic kidney disease, high concentrations of calcium and phosphate induce mineral stress, which causes a reduction of calcification inhibitors secreted by exosomes[39].
Elevated levels of calcium induced sphingomyelin phosphodiesterase 3 (SMPD3) expression and secretion of VSMC-derived exosomes, while chemical inhibition of SMPD3 inhibited the calcification process[39]. Exosomes contain calcium and are rich in calcium-binding annexins [Table 1], so during calcification, exosomes act as nucleation sites for the formation of HA deposits through binding of two exosomal membrane proteins: annexin A6 and phosphatidylserine (PS)[38]. Aggregation of EVs, through annexins on their surface, and their tethering to ECM components actively enhance their mineralization capacity[105,106]. Recently, senescence has been implicated in promoting the nucleation of EVs as a study showed EVs from senescent cells were high in calcium and contained increased levels of annexin A2, annexin A6, and BMP2[12,107].
Role of EVs and their cargo in calcification, amyloidosis and ECM aging
| Role of EVs | EV cargo | Ref. | |
| Vascular calcification | Nucleus for mineral growth | Annexin A1, annexin 2, annexin A5, annexin A6, BMP2, TNAP, phosphatidylserine, hydroxyapatite, Grp78, collagen I | [38,39,105-108] |
| Amyloidosis | Cleavage of amyloid proteins | APP, PSEN1, BACE1 | [91,109] |
| Secretion of amyloidogenic proteins and peptides | Medin, MFGE8, amyloid-β, tau, prion protein, α-synuclein, PMEL, SOD1 | [37,110-113] | |
| Promotion and acceleration of amyloid aggregation | Apolipoprotein E, HSPG2, cellular prion protein, cholesterol, glycosphingolipids | [37,114-116] | |
| Facilitating uptake of amyloid seeds by neighboring cells | Phosphatidylserine | [116] | |
| ECM aging | Degradation of ECM components | MMP2, MMP14, ADAM9, ADAM10, ADAMTS5 | [117-119] |
| Collagen crosslinking | LOXL2, PLOD1, PLOD2, PLOD3, P3H1, TGM2 | [119,120] |
In addition to exosomes, other types of EVs have been shown to be involved in calcification; PS in apoptotic bodies helped the formation of calcium phosphate, providing a nucleation site for mineral growth[51]. In addition, a study found microvesicles extracted from the plasma of a group of elderly people had higher concentrations of annexin (which may act as possible nucleation sites), promoting vascular calcification; this correlated with high concentrations of calcium and increased quantities of bone-related proteins[107].
EVs may also contain miRNAs, small endogenous molecules that regulate mRNA transcription repression through binding to the UTR[121,122]. Altered levels of pro-calcific miRNAs contained in VSMC-derived calcified exosomes in the extracellular space have been shown to contribute to the pro-osteogenic phenotype of VSMCs[123]. Furthermore, many miRNAs identified in exosomes can regulate Runx2 and Smad1 gene expression[124]. Together, these studies demonstrate a key role for EVs in vascular calcification through several distinct mechanisms.
EVs in amyloidosis
Recently, there has been growing interest in the cargo of EVs, particularly in the context of amyloid-related pathologies. Exosomes, sEVs, and MVEs have also been identified as mediators in physiological and age-related pathological amyloid metabolism[40,125-129]. Several amyloid proteins have been identified in EVs, including Aβ, medin, tau, and α-synuclein [Table 1]. To date, research has focussed mainly on the role of EVs in neurodegenerative diseases. For example, prion proteins (PrP), which cause transmissible spongiform encephalopathies, have been associated with exosomes from neurons, epithelial cells, and neuroglial cells, and increased exosome secretion was shown to increase the infectiousness of abnormal prion proteins (PrPSc)[110,114,130]. Exosomes from neurons and cerebral spinal fluid can also contain α-synuclein, a component of Lewy bodies, which is a characteristic of Parkinson’s disease and dementia with Lewy bodies[111,131].
Recent research has shown that VSMC-derived sEVs drive aggregation and accumulation of medin amyloid[37]. sEVs were shown to deposit small medin aggregates into the ECM and accelerate the aggregation of medin into amyloid fibrils by acting as a nucleus for fibril growth. Senescent VSMCs secreted more sEVs and increased deposition of medin in the ECM and inhibition of sEV secretion with 3-OMS treatment attenuated this. Interestingly, while 3-OMS decreased the overall amount of medin in the senescent ECM, it could not prevent the accumulation of medin in a fibril-like form. Furthermore, super-resolution microscopy showed sEVs from senescent VSMCs were coated with large fibril-like medin seeds, suggesting senescent sEVs may be a sufficient trigger for fibril formation.
Not all amyloids are pathological, as there have been some amyloids identified that are thought to have physiological roles[132]. Exosomes have been shown to play a pivotal role in the formation of amyloid fibrils from fragments of the premelanosomal protein (PMEL) in melanocytes[133]. These fibrils act as a scaffold for the polymerization of melanin, which protects cells from UV and oxidative stress. Cleavage of PMEL into amyloidogenic fragments occurs in MVEs by β-secretase 2 (BACE2) and their aggregation into fibrils is nucleated by ILVs, which are formed in MVEs prior to secretion as exosomes[129,133].
A similar mechanism to PMEL processing is observed during the accumulation of Aβ in the brain in AD. Numerous in vivo studies have shown that circulating exosomes in AD mouse models and AD patient body fluids, including blood and CSF, contain the C-terminal fragments of amyloid precursor protein (APP)[109] and soluble Aβ42[134,135]. Processing of APP resulting in amyloid production occurs predominantly in the endosomal pathway, while non-amyloidogenic APP processing occurs at the cell surface membrane[125]. Aβ peptides are formed from cleavage of the membrane APP by β-secretase 1 (BACE1) in MVEs, followed by cleavage of the membrane-anchored C-terminal fragment by γ-secretase. The optimum activity of BACE1 is at acidic pH[136], explaining its dominant localization at the endosomal membrane, as endosomal compartments have acidic pH (6.1-6.8)[137]. Aβ peptides are sorted in MVEs and are loaded into the ILVs, which are then released as exosomes[92]. Following secretion, exosomes could function as a nucleation site for amyloid plaque formation[92,138]. The release of Aβ into the extracellular space could also infect neighboring cells and act as amyloid seeds to accelerate the progression of disease[138,139]. Interestingly, increasing exosome secretion from neuronal cells has also been described as beneficial, with a study showing Aβ-laden exosomes from neurons were taken up by microglia for degradation. Despite exosomes significantly promoting amyloidosis, there may also be a role for them in Aβ clearance which is a method for AD treatment that needs further investigation[116].
EVs in senescence
Cellular senescence triggers changes in EV secretion and cargo loading, which can propagate aging processes through paracrine and endocrine signaling, driving inflammation, gene expression, and metabolic changes in recipient cells[140]. Senescent cell-derived EVs can carry nuclear and mitochondrial DNA which activate immune signaling, such as the cyclin GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway, or toll-like receptor 9 (TLR9) mediated inflammatory response[141]. EV secretion was increased from senescent VSMCs and senescent VSMC-derived sEVs accelerated medin aggregation into fibrils through changes in sEV cargo and altered binding to HSPG2 in the ECM[37,93]. Senescent endothelial cell-derived EVs are enriched in annexins A2 and A6, BMP2, and calcium and can promote calcification of VSMCs[107]. Just as "senescent EVs" induce an aging phenotype and enhance both calcification and amyloidosis, EVs from young cells can improve an aging phenotype. EVs from young cardiac progenitor cells reversed cell senescence in vitro and in vivo through telomere elongation, inhibition of the DNA damage response, and reduced inflammation in cultured human fibroblasts and cartilage-derived stem/progenitor cells in a naturally aged rat model[142]. Previous studies have also demonstrated the role of several miRNAs, including miR-34a and miR-126b, in mediating the effects of young EVs and highlighted the potential of EVs as a novel treatment for aging pathologies[143,144].
EVs have been shown to regulate vascular calcification, amyloidosis, and cellular senescence, indicating a possible link between these aging pathologies and their contribution to vascular stiffening and CVD. The role of EV cargo, their interaction with the extracellular environment, and mechanisms regulating their secretion and accumulation need clarification.
EXTRACELLULAR MATRIX AGING AND EVS
Both calcification and amyloidosis accumulate in the extracellular space in the vessel wall, regulated in part by the secretion of EVs. The ECM is a key component of the vascular wall and is essential for the maintenance of cellular homeostasis, acting as a scaffold and supporting cellular organization in many tissues and organs. It is comprised of several families of proteins, such as collagens, proteoglycans (PGs), and MMPs, which can influence its adherent, morphological, and phenotypical characteristics. The ratio of collagens, PGs, and elastin is essential to preserve its mechanical properties. The ECM is considered actively dynamic since it frequently undergoes remodeling by proteases, in both physiological and pathological events. The remodeling process is predominantly controlled by MMPs and their inhibitors, which regulate the balance between matrix degradation and production[145]. Dysregulation of this balance is typical of aging pathologies, such as fibrosis[146], and therefore, might be implicated in cerebrovascular amyloidosis and calcification. EVs directly impact the ECM and contribute to deposition and remodeling in both physiological and pathological environments. Through their surface-bound enzymes, which include several MMPs, elastases, and collagen crosslinking enzymes, EVs can actively degrade the ECM, resulting in the turnover of matrix components [Table 1][117,119].
Vascular aging is strongly associated with ECM aging, as matrix stiffness is the result of a series of processes occurring with age, with major changes to the mechanical forces affecting cellular function. For instance, when elastin starts to lose its elasticity, the mechanical force stress is transferred to collagen and fibronectin which is less elastic, hindering the function of the ECM, reducing its stability, and affecting cell interactions. Hence, this process appears deleterious, because when matrix begins to stiffen, it also activates downstream molecular pathways, which enhance apoptosis[147]. A recent study showed VSMC senescence to be a key regulator of increased ECM stiffness, through the TF Sox9[119]. Changes in the composition and stiffness of the senescent ECM were able to induce a senescent phenotype in young VSMCs, including cell cycle arrest and DNA damage, causing a positive feedback loop. An interesting aspect of this study was the role of EVs, as there was an accumulation of EVs in the ECM from senescent or Sox9 overexpressing VSMCs, compared to the young or control VSMCs, respectively. The EVs were loaded with procollagen-lysine, 2-oxoglutarate 5-dioxygenase 3 (PLOD3), an enzyme that caused increased ECM stiffness through collagen crosslinking. Previous studies into lung diseases, such as chronic obstructive pulmonary disease (COPD), identified subsets of EVs that drive ECM-specific proteolysis by MMPs and elastases, severe enough to cause loss of alveolar units[148,149]. EVs from subjects with COPD were also a sufficient stimulus to cause a COPD phenotype in mice primarily through surface-bound neutrophil elastase[149], further demonstrating an important relationship between the ECM and EVs with aging and disease.
Several pathological mechanisms occur during ECM aging. Collagen crosslinking, the formation of advanced glycation end products (AGEs), and the deposition of lipids and calcium are the most studied. Interestingly, these modifications have also been detected during both arterial calcification[150-153] and amyloidosis[154,155].
ECM is known to have a role in bone formation and remodeling[83] beyond its crucial signaling function[156]. Interestingly, elastin degradation by matrix proteolytic enzymes during aging and CKD leads to vascular calcification and VSMCs switch to a synthetic phenotype[86]. MMPs, such as MMP2 and MMP9, are considered calcification enhancers during senescence. Indeed, those enzymes upregulate BMP2, promoting VSMC calcification and degradation of other key ECM components of the vascular tunica media such as elastin, fibronectin, and collagens[84,85].
Changes in VSMC-derived ECM and basement membrane components caused by senescence have been described in AMA, where senescence increased HSPG2 expression and extracellular deposition, increasing medin accumulation and aggregation[37]. Knock-down of HSPG2 in the senescent ECM attenuated medin fibril formation and deposition. Interestingly, in this study, decreasing sEV secretion decreased the accumulation of medin in the senescent ECM, but did not decrease the deposition of medin in a fibrillar form. Additionally, sEVs from senescent VSMCs contained larger medin fibril seeds and preferentially bound to HSPG2 in the ECM, indicating a complex relationship between cellular senescence, sEV secretion, and changes in ECM composition.
Changes in ECM composition have also been described in many neurodegenerative diseases, such as AD and CAA. Thickening and delamination of ECM and basement membrane have been shown to be present with greater severity in the AD brain[157]. Data suggest that a change in ECM composition of aged brains creates an amyloidogenic environment as ECM proteins, such as collagen IV and fibronectin, are increased in AD brains, with fibronectin shown to bind Aβ and augment amyloid deposition in vivo[158].
TOXICITY OF CALCIFICATION AND AMYLOID
Both calcification and amyloid have been shown to have toxic effects on the vasculature. Previous studies have described the toxicity of HA nanoparticles on VSMCs, which reduced cell viability through disruption of cell membranes, increased reactive oxygen species production, and mitochondrial dysfunction, resulting in cell necrosis[159]. Uptake of HA particles by VSMCs was also shown to induce expression of osteogenic genes, such as BMP2, Runx2, and ALP, and induced cell death, causing accumulation of apoptotic bodies, indicating a positive feedback cycle where calcification exacerbates calcification. HA crystals in the vessel can also be taken up by macrophages, causing the secretion of several proinflammatory cytokines, including TNFα, IL-1β, and IL-8, mediated by protein kinase C alpha (PKCα)[160,161]. Inflammation is a hallmark of atherosclerosis, intimal and medial calcification and can promote VSMC apoptosis or osteogenic differentiation, again demonstrating how HA accumulation can exacerbate calcification.
The formation of amyloid fibrils from oligomeric aggregates has previously been discussed as a protective mechanism[24,162,163]. Small, pre-fibrillar aggregates are toxic to cells, whereas mature amyloid fibrils, the precursor protein or monomeric form may not[164]. A possible mechanism for this is the exposure of residues normally embedded inside the protein being exposed to and interacting with cell membranes, intracellular and extracellular components[24,165,166]. In Aβ pathology, the accumulation of intracellular or extracellular oligomeric aggregates precedes the formation of extracellular plaques and contributes to the main pathological effects of AD[167]. Similarly, previous research into the effects of medin showed that medin aggregates induced several pathological effects, such as endothelial dysfunction, vascular inflammation, and VSMC death[168,169]. Acceleration of medin fibril formation by heparin decreased the toxic effects of medin, suggesting the formation of mature, benign amyloid fibrils in AMA could be a protective mechanism against the toxic nature of smaller medin aggregates[170].
Both calcification and amyloidosis can contribute to ECM stiffening. HA accumulation and ECM remodeling during calcification both directly impact arterial stiffening, through increased turnover of collagen and degradation of elastin, causing weakening of the ECM[171,172]. In addition, medial calcification is associated with increased PWV and increased pulse pressure, which are linked to the accelerated development of CVD[22]. The pathological nature of amyloid is more complex as research has shown that fibrous amyloid deposits can contribute to reduced elasticity of vessels[65]. However, a previous study showing AMA was associated with weakening of the aortic wall in the development of thoracic aortic aneurysm, demonstrated the main pathological effects of medin can be attributed to toxic pre-fibrillar aggregates[169]. This suggests that toxic aggregates could contribute to vascular pathologies, which may then be exacerbated by diminished elasticity and vessel wall weakening by amyloid fibrils.
CO-OCCURRENCE OF CALCIFICATION AND AMYLOID
Even though vascular calcification and amyloidosis are age-related pathologies occurring in patients over the age of 60, there have been few systematic studies investigating their co-occurrence[12-14]. In other disease contexts, localized amyloid deposits have been associated with calcification in skin, larynx, and lungs, forming calcified tumoral amyloids, known as calcified amyloidomas[173-175].
Amyloid deposits in the cardiovascular system have been observed in calcified regions of the coronary artery and aortic valves and have been associated with aortic stenosis and calcific aortic valve disease (CAVD)[176]. Recent studies have suggested that amyloid deposition might play a direct role in the pathogenesis of CAVD. Analysis of the morphology of calcified valves described circular cavities within mineralized structures, filled with fibrous protein, which exhibited birefringence, indicating the presence of amyloid at the core[177]. A study showed 88% of aortic valves from CAVD patients had amyloid accumulation, with all the patients over 50 years having amyloid deposition[178]. Interestingly, only one-third of subjects under 30 years had amyloid deposits, indicating a potential link with aging, CAVD, and amyloid. Immunohistochemical staining was used to try to identify the amyloid protein involved, with apoA-I being detected in the mineralized areas of the valves. In vitro treatment of valve interstitial cells with amyloid extracts from calcified valves enhanced mineralization and apoptosis[177]. These studies highlighted the potential effect of apoA-I amyloid as an "interface" between the organic and mineral phases in CAVD. A previous study identified sEVs, described as matrix vesicles (MVs), as involved in CAVD, with annexin-rich MVs detected in calcified human aortic valves[179]. An interesting aspect of CAVD to investigate further would be the mechanisms regulating the accumulation of both mineral and amyloid in the valve to identify common features, such as EVs, and identify novel therapeutic targets.
In the early 1960s, Ratinov first used ex vivo radiography and microscopic analysis to reveal the presence of intracranial arterial calcification (IAC) in the extradural region of human internal carotid arteries[180]. IAC has been documented in many Caucasian and Asian populations and was shown to be a major element of cerebral atherosclerosis[181-183]. Recently, IAC has gained scientific interest as it is thought to be a significant cause of ischemic stroke[184]. IAC shares some common risk factors with amyloidosis and other cardiovascular diseases. Aging appears to be a major risk factor for IAC; an early study on the pathology did not observe any sign of IAC in patients who were 20 to 30 years old. Moreover, further research has demonstrated that IAC clinical outcomes worsen in concomitance with increasing age[181,182,185]. In addition, the imbalance of calcium and phosphate levels can be considered a risk factor as a high percentage of IAC cases have been recorded in patients with CKD[186-188].
Recent investigations have brought to light a significant association between a hereditary variant of CAA, known as the Dutch type, and IAC[189]. Studies have revealed a connection between osteopontin (OPN), and the TGFβ1/phospho-SMAD2/3 signaling pathway in calcified arteries affected by hereditary CAA[190,191]. Interestingly, the researchers observed a correlation between the pro-calcific accumulation of collagen 1, OPN, and the activation of the pSMAD2/3 pathway with the severity of CAA. This observation underscored the vital role of the ECM in calcified vessels impacted by hereditary CAA[189]. Based on their findings and the spatial relationship between vascular calcification and the deposition of Aβ in the brain vessel medial layer, the researchers proposed that calcification is more likely a characteristic of advanced-stage CAA rather than a cause of amyloid deposition.
Investigations into hereditary CAA cases evidenced a consistent involvement of basal ganglia vessels in IAC[192]. The calcification patterns observed in vessels affected by hereditary CAA are categorized as a subtype of microvasculopathies associated with CAA, presenting as a secondary manifestation of the disease[189]. Nonetheless, unraveling the underlying mechanisms that underpin the calcification of certain vessels in the context of CAA remains a mystery that warrants further exploration.
COMMON AND RECIPROCAL MECHANISMS OF CALCIFICATION AND AMYLOIDOSIS
The role of EVs in calcification and amyloidosis in the vasculature is interesting, as VSMC-derived sEVs and exosomes have been implicated in the deposition and nucleation of both HA and amyloid. The precise mechanisms governing what initiates EVs to mineralize, aggregate protein, or both are unknown, but it is clear that EVs are an important trigger for both pathologies and could account for their concomitant development in the vessel wall with age.
As for the reciprocal effects of calcification and amyloid, evidence suggests amyloid could enhance vascular calcification through different mechanisms. Oligomers can disrupt cell membranes and create pores, which can cause mineral ion imbalance, and oligomeric forms of medin have been shown to be toxic to cells and able to upregulate the activity of MMP2[168,169]. Apoptosis can induce vascular calcification by the formation of apoptotic bodies, which can act as nucleation sites for mineral formation[51]. Previous research has shown that MMPs can also enhance vascular calcification through upregulation of BMP2 and ECM degradation[85,86,193]. Therefore, extracellular deposition of medin aggregates, which are increased with aging, could exacerbate vascular calcification.
Amyloid fibrils display distinct stereochemical properties, for example, the presence of charged residues along the fibril surface, which could enhance its capacity for binding calcium and phosphate and nucleating mineral growth[176]. This is demonstrated in cases such as mineralization of tooth enamel, where amyloid-like structures of amelogenin form a nidus for mineralization of calcium-phosphate mineral[194]. HSPGs, including HSPG2, are common components of amyloid plaques and have been shown to directly enhance the aggregation of amyloid proteins, such as medin[37]. HSPGs are also known to bind calcium, providing another scaffold for mineralization with amyloid deposits.
Conversely, while evidence suggests conditions that promote calcification, such as mineral stress, could also promote amyloid formation through ECM changes and EV secretion, no studies have described a direct effect of mineralization on amyloidosis. Both amyloid and HA formation are nucleation-dependent mechanisms, with evidence showing amyloid could nucleate mineral growth, yet any effect of HA or mineral deposits on enhancing amyloid growth has not been discussed.
FUTURE DIRECTIONS FOR VASCULAR AGING RESEARCH
There are many questions remaining regarding the accumulation of HA and amyloid with age and if there are connections between the processes. While there is strong evidence for an association between pathologies, direct causal links, particularly in the vasculature, are not understood. From studies into CAVD, IAC, and tooth enamel formation, it could be suggested that amyloid acts as a scaffold for mineral growth. In the aorta, medin amyloid and HA accumulate extracellularly with age, both regulated by VSMC-derived sEVs and exosomes; however, to date, there has been no research into a causal relationship. Therefore, investigating whether medin and other vascular amyloid proteins, such as Aβ, can nucleate minerals to drive calcification or vice versa would be valuable. Along with being a physical template for mineralization, there is evidence to indicate that amyloid could enhance vascular calcification through cellular pathways, such as oxidative stress, membrane pore formation, and ECM remodeling through increased MMP secretion. However, there is currently a lack of consensus as to what form of amyloid contributes to vascular dysfunction, as studies have discussed pathological outcomes of both oligomeric and fibrillar amyloid species. Previous studies have discussed whether aggregation of toxic oligomers into “benign” fibrils is a protective mechanism, in which case therapies to prevent amyloid formation may be detrimental to vascular function. Further investigation is therefore needed to understand the contribution of different amyloid species during vascular aging and in vascular calcification.
Identification of therapeutic targets of amyloidosis and calcification could attenuate vascular stiffening with age, which is known to contribute to CVD morbidity and mortality. In recent years, there has been a huge push towards high throughput screening (HTS) as a platform to test thousands of drugs in an in vitro setting. To date, there has been limited use of HTS to identify novel drugs and targets for calcification. However, HTS has been used to identify compounds that affect amyloid formation, particularly Aβ, α-synuclein, and TTR[195]. In the past, small compounds that could block Aβ aggregation could be counteracted by the presence of a lipid membrane, but a recent study identified compounds that were effective with vesicles present to confirm inhibition of amyloid formation at the membrane interface[196]. Additionally, as previously discussed, the effects of blocking amyloid fibril formation may also result in cellular dysfunction caused by small aggregates. These are two important aspects of HTS that should be considered in the future for identification of potential therapies for amyloidosis.
VSMC senescence has been implicated in calcification and vascular amyloidosis and drugs that induce apoptosis of senescent cells, senolytics, have been shown to be effective in AD mouse models, with Aβ or tau pathology[88,197]. There is less known about the efficacy of senolytics in calcification, although one study showed that treatment with Dasatinib + Quercetin caused clearance of senescent cells in the medial layer and reduced aortic calcification in both aged and hypercholesterolaemic mice[198]. This was associated with a reduction in osteogenic gene expression and significantly improved vasomotor function. Therefore, senolytics may be a potential therapeutic intervention for both vascular calcification and amyloidosis.
In conjunction with senescence, sEVs are mediators and potential initial triggers for both HA and amyloid accumulation in the vasculature. There has been some research into targeting EV secretion to alleviate both pathologies in both in vitro and in vivo studies. In a mouse model of CKD, inhibition of sEV secretion with a neutral sphingomyelinase inhibitor, GW4869, resulted in reduced aortic calcification and osteogenic differentiation of VSMCs[123]. Blocking sEV secretion also attenuated the calcification of human and mouse VSMCs in vitro, under calcifying or diabetic conditions[199]. In AD mouse models, GW4869 significantly reduced Aβ plaque load in the brain[135,200]. These studies highlight not only the therapeutic potential of targeting sEV secretion but also the importance of sEVs in the initiation and development of calcification and amyloidosis.
CONCLUSION
This review has highlighted that amyloid and HA deposition in the vasculature share mechanistic processes and display synergies, which may account for their co-localization in the vessel wall [Figure 5]. Mineral stress and cellular senescence can increase EV production and promote osteogenic changes in VSMCs, which could also promote amyloid deposition, while amyloid in both oligomeric and fibrillar forms could nucleate minerals and enhance calcification. Importantly, there is evidence showing EV secretion, senescence, and ECM modifications can enhance both pathologies, suggesting therapies that target these common mechanisms may alleviate age-associated vascular stiffening.
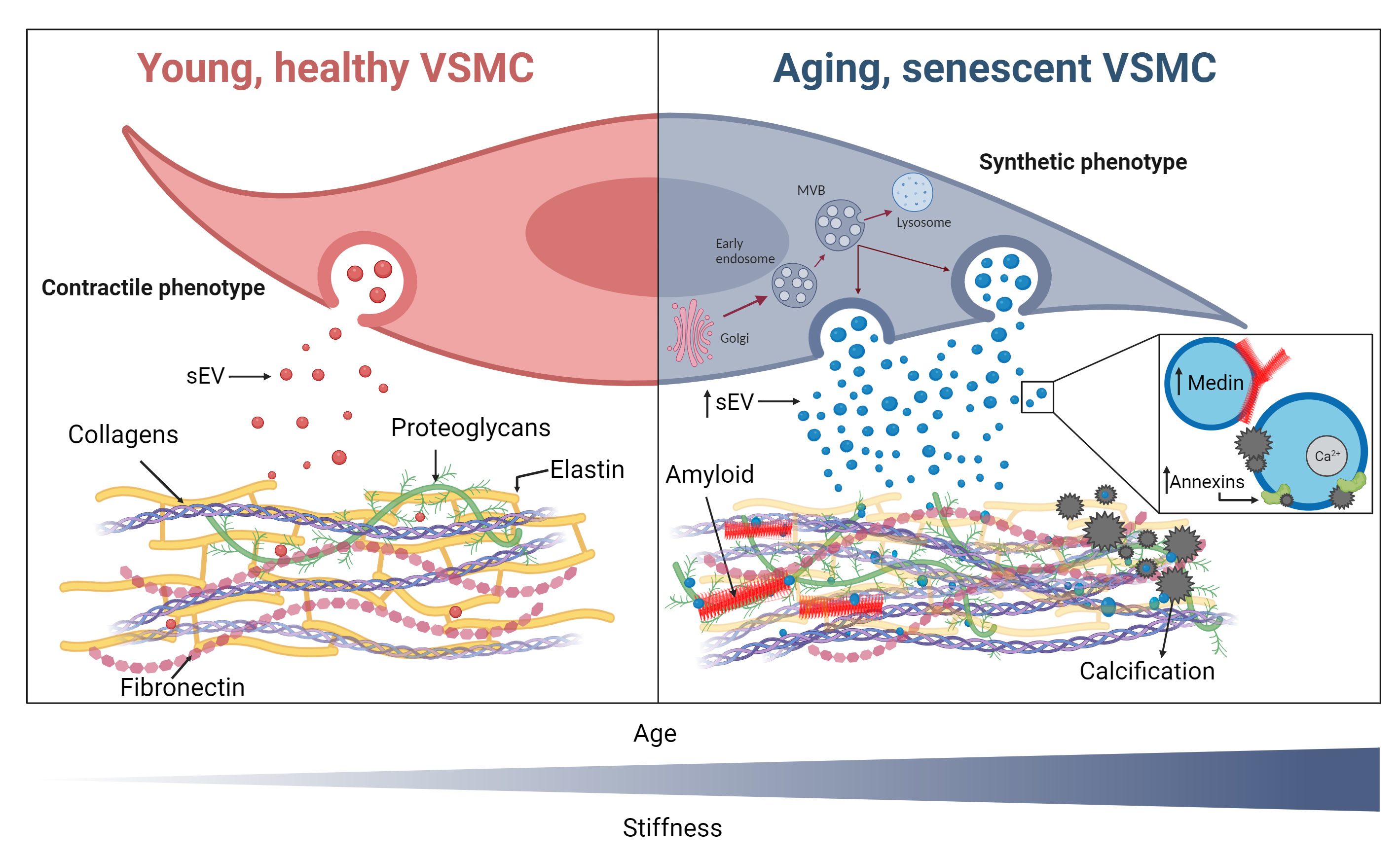
Figure 5. Young, healthy vascular smooth muscle cells (VSMCs) have a contractile phenotype and a healthy extracellular matrix (ECM), which undergoes turnover and remodeling to maintain its compliance and regulate vascular tone and integrity. VSMCs undergoing senescence alter the composition and stiffness of the ECM, increasing collagen and fibronectin deposition and decreasing elastin. Senescent VSMCs secrete more small extracellular vesicles (sEVs), which promote both calcification and amyloid aggregation and deposition. There is increased endosomal activity and formation of multivesicular endosomes (MVE) which contain sEVs that are degraded by lysosomes or released by binding to the membrane. sEVs from senescent VSMCs contain more medin in a fibrillar form and can accelerate its accumulation as fibrils in the ECM. Senescent sEVs also contain more calcium and annexins, which enhance mineralization. What triggers sEVs to either mineralize or aggregate amyloid is unknown. (Created with BioRender.com)
DECLARATIONS
Authors’ contributions
Conceptualization, writing and editing : Whitehead M, Antonazzi M, Shanahan CM
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by a Grant from the British Heart Foundation to C.S. BHF: RG/F/21/110064, https://researchgrants.bhf.org.uk. The funding body had no role in the experiment design, collection, analysis, and interpretation of data, and writing.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024.
REFERENCES
1. Lakatta EG. So! What′s aging? Is cardiovascular aging a disease? J Mol Cell Cardiol 2015;83:1-13.
2. Nichols M, Townsen N, Scarborough P, et al. European cardiovascular disease statistics 2012 editon. Avaliable from: https://www.escardio.org/static-file/Escardio/Press-media/press-releases/2013/EU-cardiovascular-disease-statistics-2012.pdf [Last accessed on 22 Jan 2024].
3. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res 2016;118:692-702.
4. Reynolds JL, Joannides AJ, Skepper JN, et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol 2004;15:2857-67.
5. Brunner EJ, Shipley MJ, Witte DR, et al. Arterial stiffness, physical function, and functional limitation: the Whitehall II study. Hypertension 2011;57:1003-9.
6. Kim ED, Tanaka H, Ballew SH, et al. Associations between kidney disease measures and regional pulse wave velocity in a large community-based cohort: the atherosclerosis risk in communities (ARIC) study. Am J Kidney Dis 2018;72:682-90.
7. Wehrum T, Dragonu I, Strecker C, et al. Influence of pulse wave velocity on atherosclerosis and blood flow reversal in the aorta: a 4-dimensional flow magnetic resonance imaging study in acute stroke patients and matched controls. J Thorac Imaging 2022;37:42-8.
8. Hughes TM, Kuller LH, Barinas-Mitchell EJ, et al. Pulse wave velocity is associated with β-amyloid deposition in the brains of very elderly adults. Neurology 2013;81:1711-8.
9. Rouch L, Cestac P, Sallerin B, et al. Pulse wave velocity is associated with greater risk of dementia in mild cognitive impairment patients. Hypertension 2018;72:1109-16.
10. Laurent S, Boutouyrie P, Asmar R, et al. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension 2001;37:1236-41.
11. Sutton-Tyrrell K, Najjar SS, Boudreau RM, et al. Health ABC Study. Elevated aortic pulse wave velocity, a marker of arterial stiffness, predicts cardiovascular events in well-functioning older adults. Circulation 2005;111:3384-90.
12. Mas-Bargues C, Borrás C, Alique M. The contribution of extracellular vesicles from senescent endothelial and vascular smooth muscle cells to vascular calcification. Front Cardiovasc Med 2022;9:854726.
13. Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm 2002;109:813-36.
14. Itoh Y, Yamada M, Hayakawa M, Otomo E, Miyatake T. Cerebral amyloid angiopathy: a significant cause of cerebellar as well as lobar cerebral hemorrhage in the elderly. J Neurol Sci 1993;116:135-41.
15. Davies HA, Caamaño-Gutiérrez E, Chim YH, et al. Idiopathic degenerative thoracic aneurysms are associated with increased aortic medial amyloid. Amyloid 2019;26:148-55.
16. Abedin M, Tintut Y, Demer LL. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol 2004;24:1161-70.
17. McClelland RL, Chung H, Detrano R, Post W, Kronmal RA. Distribution of coronary artery calcium by race, gender, and age: results from the multi-ethnic study of atherosclerosis (MESA). Circulation 2006;113:30-7.
18. Shaw LJ, Raggi P, Berman DS, Callister TQ. Coronary artery calcium as a measure of biologic age. Atherosclerosis 2006;188:112-9.
19. Jeziorska M, McCollum C, Wooley DE. Observations on bone formation and remodelling in advanced atherosclerotic lesions of human carotid arteries. Virchows Arch 1998;433:559-65.
20. Mackey RH, Venkitachalam L, Sutton-tyrrell K. Calcifications, arterial stiffness and atherosclerosis. Adv Cardiol 2007;44:234-244.
21. Doherty TM, Fitzpatrick LA, Inoue D, et al. Molecular, endocrine, and genetic mechanisms of arterial calcification. Endocr Rev 2004;25:629-72.
22. London GM, Guérin AP, Marchais SJ, Métivier F, Pannier B, Adda H. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant 2003;18:1731-40.
23. Niskanen L, Siitonen O, Suhonen M, Uusitupa MI. Medial artery calcification predicts cardiovascular mortality in patients with NIDDM. Diabetes Care 1994;17:1252-6.
24. Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 2006;75:333-66.
25. Wang Y, Feng X, Shen B, Ma J, Zhao W. Is vascular amyloidosis intertwined with arterial aging, hypertension and atherosclerosis? Front Genet 2017;8:126.
26. Cornwell GG 3rd, Johnson KH, Westermark P. The age related amyloids: a growing family of unique biochemical substances. J Clin Pathol 1995;48:984-9.
27. Cornwell GG 3rd, Westermark P. Senile amyloidosis: a protean manifestation of the aging process. J Clin Pathol 1980;33:1146-52.
28. Mucchiano G, Cornwell GG 3rd, Westermark P. Senile aortic amyloid. Evidence for two distinct forms of localized deposits. Am J Pathol 1992;140:871-7.
29. Galant NJ, Westermark P, Higaki JN, Chakrabartty A. Transthyretin amyloidosis: an under-recognized neuropathy and cardiomyopathy. Clin Sci 2017;131:395-409.
30. Haggqvist B, Naslund J, Sletten K, et al. Medin: an integral fragment of aortic smooth muscle cell-produced lactadherin forms the most common human amyloid. Proc Natl Acad Sci USA 1999;96:8669-74.
31. Larsson A, Peng S, Persson H, et al. Lactadherin binds to elastin - a starting point for medin amyloid formation? Amyloid 2006;13:78-85.
32. Weller RO, Boche D, Nicoll JA. Microvasculature changes and cerebral amyloid angiopathy in Alzheimer’s disease and their potential impact on therapy. Acta Neuropathol 2009;118:87-102.
33. Keable A, Fenna K, Yuen HM, et al. Deposition of amyloid β in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim Biophys Acta 2016;1862:1037-46.
34. Baker DJ, Childs BG, Durik M, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016;530:184-9.
35. Girndt M, Seibert E. Premature cardiovascular disease in chronic renal failure (CRF): a model for an advanced ageing process. Exp Gerontol 2010;45:797-800.
36. Burton DG, Matsubara H, Ikeda K. Pathophysiology of vascular calcification: pivotal role of cellular senescence in vascular smooth muscle cells. Exp Gerontol 2010;45:819-24.
37. Whitehead M, Yusoff S, Ahmad S, et al. Vascular smooth muscle cell senescence accelerates medin aggregation via small extracellular vesicle secretion and extracellular matrix reorganization. Aging Cell 2023;22:e13746.
38. Kapustin AN, Shanahan CM. Emerging roles for vascular smooth muscle cell exosomes in calcification and coagulation. J Physiol 2016;594:2905-14.
39. Kapustin AN, Chatrou ML, Drozdov I, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res 2015;116:1312-23.
40. van Niel G. Study of exosomes shed new light on physiology of amyloidogenesis. Cell Mol Neurobiol 2016;36:327-42.
41. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 2004;84:767-801.
42. Rudijanto A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Med Indones 2007;39:86-93. Avaliable from: http://inaactamedica.org/archives/2007/17933075.pdf [Last accessed on 22 Jan 2024].
43. Steucke KE, Tracy PV, Hald ES, Hall JL, Alford PW. Vascular smooth muscle cell functional contractility depends on extracellular mechanical properties. J Biomech 2015;48:3044-51.
44. Kapustin A, Tsakali SS, Whitehead M, et al. Extracellular vesicles stimulate smooth muscle cell migration by presenting collagen VI. bioRxiv 2023;1:551257.
45. Goodman WG, Goldin J, Kuizon BD, et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med 2000;342:1478-83.
46. Bi W, Huang W, Whitworth DJ, et al. Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc Natl Acad Sci USA 2001;98:6698-703.
47. Wilkie AO, Tang Z, Elanko N, et al. Functional haploinsufficiency of the human homeobox gene MSX2 causes defects in skull ossification. Nat Genet 2000;24:387-90.
48. Komori T. Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res 2010;339:189-95.
49. Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res 2011;109:697-711.
50. Ueland T, Dahl CP, Gullestad L, et al. Circulating levels of non-phosphorylated undercarboxylated matrix Gla protein are associated with disease severity in patients with chronic heart failure. Clin Sci 2011;121:119-27.
51. Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res 2000;87:1055-62.
52. Naiki H, Hashimoto N, Suzuki S, Kimura H, Nakakuki K, Gejyo F. Establishment of a kinetic model of dialysis-related amyloid fibril extension in vitro. Amyloid 1997;4:223-32.
53. Serio TR, Cashikar AG, Kowal AS, et al. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 2000;289:1317-21.
54. MacRaild CA, Stewart CR, Mok YF, et al. Non-fibrillar components of amyloid deposits mediate the self-association and tangling of amyloid fibrils. J Biol Chem 2004;279:21038-45.
55. Peng S, Glennert J, Westermark P. Medin-amyloid: a recently characterized age-associated arterial amyloid form affects mainly arteries in the upper part of the body. Amyloid 2005;12:96-102.
56. Miura Y, Tsumoto H, Iwamoto M, et al. Age-associated proteomic alterations in human aortic media. Geriatr Gerontol Int 2019;19:1054-62.
57. Cheng M, Li BY, Li XL, et al. Correlation between serum lactadherin and pulse wave velocity and cardiovascular risk factors in elderly patients with type 2 diabetes mellitus. Diabetes Res Clin Pract 2012;95:125-31.
58. Wang M, Fu Z, Wu J, et al. MFG-E8 activates proliferation of vascular smooth muscle cells via integrin signaling. Aging Cell 2012;11:500-8.
59. Chiang HY, Chu PH, Chen SC, Lee TH. MFG-E8 promotes osteogenic transdifferentiation of smooth muscle cells and vascular calcification by regulating TGF-β1 signaling. Commun Biol 2022;5:364.
60. Chiang HY, Chu PH, Lee TH. MFG-E8 mediates arterial aging by promoting the proinflammatory phenotype of vascular smooth muscle cells. J Biomed Sci 2019;26:61.
61. Peng S, Westermark GT, Näslund J, Häggqvist B, Glennert J, Westermark P. Medin and medin-amyloid in ageing inflamed and non-inflamed temporal arteries. J Pathol 2002;196:91-6.
62. Coria F, Castano EM, Frangione B. Brain amyloid in normal aging and cerebral amyloid angiopathy is antigenically related to Alzheimer’s disease beta-protein. Am J Pathol 1987;129:422-8.
63. Kawai M, Kalaria RN, Cras P, et al. Degeneration of vascular muscle cells in cerebral amyloid angiopathy of Alzheimer disease. Brain Res 1993;623:142-6.
64. Attems J. Sporadic cerebral amyloid angiopathy: pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol 2005;110:345-59.
65. Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP Jr. Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 1991;30:637-49.
66. De Meyer GR, De Cleen DM, Cooper S, et al. Platelet phagocytosis and processing of β-amyloid precursor protein as a mechanism of macrophage activation in atherosclerosis. Circ Res 2002;90:1197-204.
67. Kokjohn TA, Van Vickle GD, Maarouf CL, et al. Chemical characterization of pro-inflammatory amyloid-β peptides in human atherosclerotic lesions and platelets. Biochim Biophys Acta 2011;1812:1508-14.
68. Stamatelopoulos K, Sibbing D, Rallidis LS, et al. Amyloid-β (1-40) and the risk of death from cardiovascular causes in patients with coronary heart disease. J Am Coll Cardiol 2015;65:904-16.
69. Stamatelopoulos K, Stellos K. Circulating amyloid-β (1-40) predicts clinical outcomes in patients with heart failure. Rev Esp Cardiol 2017;70:905-6.
70. Erusalimsky JD. Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol 2009;106:326-32.
71. Celermajer DS, Sorensen KE, Spiegelhalter DJ, Georgakopoulos D, Robinson J, Deanfield JE. Aging is associated with endothelial dysfunction in healthy men years before the age-related decline in women. J Am Coll Cardiol 1994;24:471-6.
72. Nakano-Kurimoto R, Ikeda K, Uraoka M, et al. Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am J Physiol Heart Circ Physiol 2009;297:H1673-84.
73. Liu Y, Drozdov I, Shroff R, Beltran LE, Shanahan CM. Prelamin A accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ Res 2013;112:e99-109.
74. Lim K, Lu TS, Molostvov G, et al. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation 2012;125:2243-55.
75. Hu MC, Shi M, Zhang J, et al. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol 2011;22:124-36.
76. Kovacic JC, Moreno P, Nabel EG, Hachinski V, Fuster V. Cellular senescence, vascular disease, and aging: part 2 of a 2-part review: clinical vascular disease in the elderly. Circulation 2011;123:1900-10.
77. Kovacic JC, Moreno P, Hachinski V, Nabel EG, Fuster V. Cellular senescence, vascular disease, and aging: part 1 of a 2-part review. Circulation 2011;123:1650-60.
78. Zickler D, Luecht C, Willy K, et al. Tumour necrosis factor-alpha in uraemic serum promotes osteoblastic transition and calcification of vascular smooth muscle cells via extracellular signal-regulated kinases and activator protein 1/c-FOS-mediated induction of interleukin 6 expression. Nephrol Dial Transplant 2018;33:574-85.
79. Grootaert MOJ, Moulis M, Roth L, et al. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc Res 2018;114:622-34.
80. Matthews C, Gorenne I, Scott S, et al. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res 2006;99:156-64.
81. Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol 2021;22:75-95.
82. Jeck WR, Siebold AP, Sharpless NE. Review: a meta-analysis of GWAS and age-associated diseases. Aging Cell 2012;11:727-31.
83. Chow AK, Cena J, Schulz R. Acute actions and novel targets of matrix metalloproteinases in the heart and vasculature. Br J Pharmacol 2007;152:189-205.
84. Chen NX, O'Neill KD, Chen X, Kiattisunthorn K, Gattone VH, Moe SM. Activation of arterial matrix metalloproteinases leads to vascular calcification in chronic kidney disease. Am J Nephrol 2011;34:211-9.
85. Zhao YG, Meng FX, Li BW, et al. Gelatinases promote calcification of vascular smooth muscle cells by up-regulating bone morphogenetic protein-2. Biochem Biophys Res Commun 2016;470:287-93.
86. Pai A, Leaf EM, El-Abbadi M, Giachelli CM. Elastin degradation and vascular smooth muscle cell phenotype change precede cell loss and arterial medial calcification in a uremic mouse model of chronic kidney disease. Am J Pathol 2011;178:764-73.
87. Ting KK, Coleman P, Kim HJ, et al. Vascular senescence and leak are features of the early breakdown of the blood-brain barrier in Alzheimer's disease models. Geroscience 2023;45:3307-31.
88. Zhang P, Kishimoto Y, Grammatikakis I, et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer's disease model. Nat Neurosci 2019;22:719-28.
90. Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011;479:232-6.
91. Rajendran L, Honsho M, Zahn TR, et al. Alzheimer's disease β-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci USA 2006;103:11172-7.
92. Rajendran L, Knobloch M, Geiger KD, et al. Increased Aβ production leads to intracellular accumulation of Aβ in flotillin-1-positive endosomes. Neurodegener Dis 2007;4:164-70.
93. Whitehead M, Antonazzi M, Shanahan CM. Senescence and extracellular vesicles: novel partners in vascular amyloidosis. Aging 2023;15:1232-4.
94. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 2013;200:373-83.
95. Simons M, Raposo G. Exosomes - vesicular carriers for intercellular communication. Curr Opin Cell Biol 2009;21:575-81.
97. Zhang J, Li S, Li L, et al. Exosome and exosomal microRNA: trafficking, sorting, and function. Genom Proteom Bioinf 2015;13:17-24.
98. Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles 2014;3:24641.
99. Muralidharan-Chari V, Clancy J, Plou C, et al. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr Biol 2009;19:1875-85.
100. Johnson SM, Dempsey C, Parker C, Mironov A, Bradley H, Saha V. Acute lymphoblastic leukaemia cells produce large extracellular vesicles containing organelles and an active cytoskeleton. J Extracell Vesicles 2017;6:1294339.
101. Ailawadi S, Wang X, Gu H, Fan GC. Pathologic function and therapeutic potential of exosomes in cardiovascular disease. Biochim Biophys Acta 2015;1852:1-11.
102. Schurgers LJ, Spronk HM, Skepper JN, et al. Post-translational modifications regulate matrix Gla protein function: importance for inhibition of vascular smooth muscle cell calcification. J Thromb Haemost 2007;5:2503-11.
103. Chen NX, Chen X, O'Neill KD, Atkinson SJ, Moe SM. RhoA/Rho kinase (ROCK) alters fetuin-A uptake and regulates calcification in bovine vascular smooth muscle cells (BVSMC). Am J Physiol Renal Physiol 2010;299:F674-80.
104. Reynolds JL, Skepper JN, McNair R, et al. Multifunctional roles for serum protein fetuin-a in inhibition of human vascular smooth muscle cell calcification. J Am Soc Nephrol 2005;16:2920-30.
105. Buffolo F, Monticone S, Camussi G, Aikawa E. Role of extracellular vesicles in the pathogenesis of vascular damage. Hypertension 2022;79:863-73.
106. Rogers MA, Buffolo F, Schlotter F, et al. Annexin A1-dependent tethering promotes extracellular vesicle aggregation revealed with single-extracellular vesicle analysis. Sci Adv 2020;6:eabb1244.
107. Alique M, Ruíz-Torres MP, Bodega G, et al. Microvesicles from the plasma of elderly subjects and from senescent endothelial cells promote vascular calcification. Aging 2017;9:778-89.
108. Furmanik M, van Gorp R, Whitehead M, et al. Endoplasmic reticulum stress mediates vascular smooth muscle cell calcification via increased release of Grp78 (Glucose-Regulated Protein, 78 kDa)-loaded extracellular vesicles. Arterioscler Thromb Vasc Biol 2021;41:898-914.
109. Laulagnier K, Javalet C, Hemming FJ, et al. Amyloid precursor protein products concentrate in a subset of exosomes specifically endocytosed by neurons. Cell Mol Life Sci 2018;75:757-73.
110. Fevrier B, Vilette D, Archer F, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci USA 2004;101:9683-8.
111. Stuendl A, Kunadt M, Kruse N, et al. Induction of α-synuclein aggregate formation by CSF exosomes from patients with Parkinson's disease and dementia with Lewy bodies. Brain 2016;139:481-94.
112. Saman S, Kim W, Raya M, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem 2012;287:3842-9.
113. Silverman JM, Christy D, Shyu CC, et al. CNS-derived extracellular vesicles from superoxide dismutase 1 (SOD1)(G93A) ALS mice originate from astrocytes and neurons and carry misfolded SOD1. J Biol Chem 2019;294:3744-59.
114. Falker C, Hartmann A, Guett I, et al. Exosomal cellular prion protein drives fibrillization of amyloid beta and counteracts amyloid beta-mediated neurotoxicity. J Neurochem 2016;137:88-100.
115. Holtzman DM. Role of apoe/Aβ interactions in the pathogenesis of Alzheimer's disease and cerebral amyloid angiopathy. J Mol Neurosci 2001;17:147-55.
116. Yuyama K, Sun H, Mitsutake S, Igarashi Y. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-β by microglia. J Biol Chem 2012;287:10977-89.
117. Patel NJ, Ashraf A, Chung EJ. Extracellular vesicles as regulators of the extracellular matrix. Bioengineering 2023;10:136.
118. Kapustin AN, Davies JD, Reynolds JL, et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res 2011;109:e1-12.
119. Faleeva M, Ahmad S, Lynham S, et al. Sox9 accelerates vascular ageing by regulating extracellular matrix composition and stiffness. Circ Res Forthcoming 2023.
120. Chen NX, O’Neill K, Chen X, Kiattisunthorn K, Gattone VH, Moe SM. Transglutaminase 2 accelerates vascular calcification in chronic kidney disease. Am J Nephrol 2013;37:191-8.
121. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 2007;9:654-9.
122. Pan W, Liang J, Tang H, et al. Differentially expressed microRNA profiles in exosomes from vascular smooth muscle cells associated with coronary artery calcification. Int J Biochem Cell Biol 2020;118:105645.
123. Koide T, Mandai S, Kitaoka R, et al. Circulating extracellular vesicle-propagated microRNA signature as a vascular calcification factor in chronic kidney disease. Circ Res 2023;132:415-31.
124. Chaturvedi P, Chen NX, O’Neill K, McClintick JN, Moe SM, Janga SC. Differential miRNA expression in cells and matrix vesicles in vascular smooth muscle cells from rats with kidney disease. PLoS One 2015;10:e0131589.
125. Rajendran L, Annaert W. Membrane trafficking pathways in Alzheimer's disease. Traffic 2012;13:759-70.
126. Rajendran L, Bali J, Barr MM, et al. Emerging roles of extracellular vesicles in the nervous system. J Neurosci 2014;34:15482-9.
127. Takahashi RH, Milner TA, Li F, et al. Intraneuronal Alzheimer Aβ42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol 2002;161:1869-79.
128. Yuyama K, Yamamoto N, Yanagisawa K. Accelerated release of exosome-associated GM1 ganglioside (GM1) by endocytic pathway abnormality: another putative pathway for GM1-induced amyloid fibril formation. J Neurochem 2008;105:217-24.
129. van Niel G, Bergam P, Di Cicco A, et al. Apolipoprotein E regulates amyloid formation within endosomes of pigment cells. Cell Rep 2015;13:43-51.
130. Guo BB, Bellingham SA, Hill AF. Stimulating the release of exosomes increases the intercellular transfer of prions. J Biol Chem 2016;291:5128-37.
131. Sackmann V, Sinha MS, Sackmann C, et al. Inhibition of nSMase2 reduces the transfer of oligomeric α-synuclein irrespective of hypoxia. Front Mol Neurosci 2019;12:200.
132. Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid - from bacteria to humans. Trends Biochem Sci 2007;32:217-24.
133. van Niel G, Charrin S, Simoes S, et al. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev Cell 2011;21:708-21.
134. Miranda AM, Lasiecka ZM, Xu Y, et al. Neuronal lysosomal dysfunction releases exosomes harboring APP C-terminal fragments and unique lipid signatures. Nat Commun 2018;9:291.
135. Dinkins MB, Dasgupta S, Wang G, Zhu G, Bieberich E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer's disease. Neurobiol Aging 2014;35:1792-800.
136. Halima S, Rajendran L. Membrane anchored and lipid raft targeted β-secretase inhibitors for Alzheimer's disease therapy. J Alzheimers Dis 2011;24 Suppl 2:143-52.
137. Maxfield FR, Yamashiro DJ. Endosome acidification and the pathways of receptor-mediated endocytosis. Adv Exp Med Biol 1987;225:189-98.
138. Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009;64:783-90.
139. Sardar Sinha M, Ansell-Schultz A, Civitelli L, et al. Alzheimer′s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol 2018;136:41-56.
140. Yin Y, Chen H, Wang Y, Zhang L, Wang X. Roles of extracellular vesicles in the aging microenvironment and age-related diseases. J Extracell Vesicles 2021;10:e12154.
141. Iske J, Seyda M, Heinbokel T, et al. Senolytics prevent mt-DNA-induced inflammation and promote the survival of aged organs following transplantation. Nat Commun 2020;11:4289.
142. Grigorian Shamagian L, Rogers RG, Luther K, et al. Rejuvenating effects of young extracellular vesicles in aged rats and in cellular models of human senescence. Sci Rep 2023;13:12240.
143. Liu Y, Liu Z, Xie Y, Zhao C, Xu J. Serum extracellular vesicles retard H9C2 cell senescence by suppressing miR-34a expression. J Cardiovasc Transl Res 2019;12:45-50.
144. Lee BR, Kim JH, Choi ES, Cho JH, Kim E. Effect of young exosomes injected in aged mice. Int J Nanomedicine 2018;13:5335-45.
145. Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci 2010;123:4195-200.
146. Decaris ML, Gatmaitan M, FlorCruz S, et al. Proteomic analysis of altered extracellular matrix turnover in bleomycin-induced pulmonary fibrosis. Mol Cell Proteomics 2014;13:1741-52.
147. Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell 2006;126:677-89.
148. Szul T, Bratcher PE, Fraser KB, et al. Toll-like receptor 4 engagement mediates prolyl endopeptidase release from airway epithelia via exosomes. Am J Respir Cell Mol Biol 2016;54:359-69.
149. Genschmer KR, Russell DW, Lal C, et al. Activated PMN exosomes: pathogenic entities causing matrix destruction and disease in the lung. Cell 2019;176:113-26.e15.
150. Brodeur MR, Bouvet C, Bouchard S, et al. Reduction of advanced-glycation end products levels and inhibition of RAGE signaling decreases rat vascular calcification induced by diabetes. PLoS One 2014;9:e85922.
151. Zhu Y, Ma WQ, Han XQ, Wang Y, Wang X, Liu NF. Advanced glycation end products accelerate calcification in VSMCs through HIF-1α/PDK4 activation and suppress glucose metabolism. Sci Rep 2018;8:13730.
152. Ren X, Shao H, Wei Q, Sun Z, Liu N. Advanced glycation end-products enhance calcification in vascular smooth muscle cells. J Int Med Res 2009;37:847-54.
153. Tanikawa T, Okada Y, Tanikawa R, Tanaka Y. Advanced glycation end products induce calcification of vascular smooth muscle cells through RAGE/p38 MAPK. J Vasc Res 2009;46:572-80.
154. Vitek MP, Bhattacharya K, Glendening JM, et al. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci USA 1994;91:4766-70.
155. Sirangelo I, Iannuzzi C. Understanding the role of protein glycation in the amyloid aggregation process. Int J Mol Sci 2021;22:6609.
156. Kim SH, Turnbull J, Guimond S. Extracellular matrix and cell signalling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor. J Endocrinol 2011;209:139-51.
157. Thomsen MS, Routhe LJ, Moos T. The vascular basement membrane in the healthy and pathological brain. J Cereb Blood Flow Metab 2017;37:3300-17.
158. Lepelletier FX, Mann DM, Robinson AC, Pinteaux E, Boutin H. Early changes in extracellular matrix in Alzheimer's disease. Neuropathol Appl Neurobiol 2017;43:167-82.
159. Huang LH, Sun XY, Ouyang JM. Shape-dependent toxicity and mineralization of hydroxyapatite nanoparticles in A7R5 aortic smooth muscle cells. Sci Rep 2019;9:18979.
160. Nadra I, Mason JC, Philippidis P, et al. Proinflammatory activation of macrophages by basic calcium phosphate crystals via protein kinase C and MAP kinase pathways: a vicious cycle of inflammation and arterial calcification? Circ Res 2005;96:1248-56.
161. Nahar-Gohad P, Gohad N, Tsai CC, Bordia R, Vyavahare N. Rat aortic smooth muscle cells cultured on hydroxyapatite differentiate into osteoblast-like cells via BMP-2-SMAD-5 pathway. Calcif Tissue Int 2015;96:359-69.
162. Pryor NE, Moss MA, Hestekin CN. Unraveling the early events of amyloid-β protein (Aβ) aggregation: techniques for the determination of Aβ aggregate size. Int J Mol Sci 2012;13:3038-72.
163. Ahmed M, Davis J, Aucoin D, et al. Structural conversion of neurotoxic amyloid-beta(1-42) oligomers to fibrils. Nat Struct Mol Biol 2010;17:561-7.
164. Kayed R, Lasagna-Reeves CA. Molecular mechanisms of amyloid oligomers toxicity. J Alzheimers Dis 2013;33 Suppl 1:S67-78.
165. Schubert D, Behl C, Lesley R, et al. Amyloid peptides are toxic via a common oxidative mechanism. Proc Natl Acad Sci USA 1995;92:1989-93.
166. Kawahara M, Kuroda Y, Arispe N, Rojas E. Alzheimer's β-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J Biol Chem 2000;275:14077-83.
167. Hu X, Crick SL, Bu G, Frieden C, Pappu RV, Lee JM. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc Natl Acad Sci USA 2009;106:20324-9.
168. Migrino RQ, Davies HA, Truran S, et al. Amyloidogenic medin induces endothelial dysfunction and vascular inflammation through the receptor for advanced glycation endproducts. Cardiovasc Res 2017;113:1389-402.
169. Peng S, Larsson A, Wassberg E, et al. Role of aggregated medin in the pathogenesis of thoracic aortic aneurysm and dissection. Lab Invest 2007;87:1195-205.
170. Madine J, Middleton DA. Comparison of aggregation enhancement and inhibition as strategies for reducing the cytotoxicity of the aortic amyloid polypeptide medin. Eur Biophys J 2010;39:1281-8.
171. Jacob MP. Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed Pharmacother 2003;57:195-202.
172. Chung AW, Yang HH, Kim JM, et al. Upregulation of matrix metalloproteinase-2 in the arterial vasculature contributes to stiffening and vasomotor dysfunction in patients with chronic kidney disease. Circulation 2009;120:792-801.
173. Rodriguez-Romero R, Vargas-Serrano B, Cortina-Moreno B, Fernandez-Gallardo JM, Cervera-Rodilla JL. Calcified amyloidoma of the larynx. AJNR Am J Neuroradiol 1996;17:1491-3. Avaliable from: https://www.ajnr.org/content/17/8/1491 [Last accessed on 22 Jan 2024].
174. Requena C, Diago A, Traves V, Llombart B, Nagore E, Sanmartin O. Cutaneous ossifying amyloidoma. Am J Dermatopathol 2022;44:760-3.
176. Sud K, Narula N, Aikawa E, et al. The contribution of amyloid deposition in the aortic valve to calcification and aortic stenosis. Nat Rev Cardiol 2023;20:418-28.
177. Audet A, Côté N, Couture C, et al. Amyloid substance within stenotic aortic valves promotes mineralization. Histopathology 2012;61:610-9.
178. Ladefoged C, Rohr N. Amyloid deposits in aortic and mitral valves. A clinicopathological investigation of material from 100 consecutive heart valve operations. Virchows Arch A Pathol Anat Histopathol 1984;404:301-12.
179. Cui L, Rashdan NA, Zhu D, et al. End stage renal disease-induced hypercalcemia may promote aortic valve calcification via Annexin VI enrichment of valve interstitial cell derived-matrix vesicles. J Cell Physiol 2017;232:2985-95.
180. Ratinov G. Extradural intracranial portion of carotid artery; a clinicopathologic study. Arch Neurol 1964;10:66-73.
181. Chen XY, Lam WW, Ng HK, Fan YH, Wong KS. The frequency and determinants of calcification in intracranial arteries in Chinese patients who underwent computed tomography examinations. Cerebrovasc Dis 2006;21:91-7.
182. Mak HK, Wong CW, Yau KK, et al. Computed tomography evaluation of intracranial atherosclerosis in Chinese patients with transient ischemic attack or minor ischemic stroke - its distribution and association with vascular risk factors. J Stroke Cerebrovasc Dis 2009;18:158-63.
183. Wu XH, Chen XY, Wang LJ, Wong KS. Intracranial artery calcification and its clinical significance. J Clin Neurol 2016;12:253-61.
184. Bugnicourt JM, Leclercq C, Chillon JM, et al. Presence of intracranial artery calcification is associated with mortality and vascular events in patients with ischemic stroke after hospital discharge: a cohort study. Stroke 2011;42:3447-53.
185. Sohn YH, Cheon HY, Jeon P, Kang SY. Clinical implication of cerebral artery calcification on brain CT. Cerebrovasc Dis 2004;18:332-7.
186. Chue CD, Townend JN, Steeds RP, Ferro CJ. Arterial stiffness in chronic kidney disease: causes and consequences. Heart 2010;96:817-23.
187. Bugnicourt JM, Chillon JM, Massy ZA, et al. High prevalence of intracranial artery calcification in stroke patients with CKD: a retrospective study. Clin J Am Soc Nephrol 2009;4:284-90.
188. Power A, Chan K, Haydar A, et al. Intracranial arterial calcification is highly prevalent in hemodialysis patients but does not associate with acute ischemic stroke. Hemodial Int 2011;15:256-63.
189. Moursel L, van der Graaf LM, Bulk M, van Roon-Mom WMC, van der Weerd L. Osteopontin and phospho-SMAD2/3 are associated with calcification of vessels in D-CAA, an hereditary cerebral amyloid angiopathy. Brain Pathol 2019;29:793-802.
190. Jian B, Narula N, Li QY, Mohler ER 3rd, Levy RJ. Progression of aortic valve stenosis: TGF-β1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg 2003;75:457-65.
191. Watson KE, Boström K, Ravindranath R, Lam T, Norton B, Demer LL. TGF-β 1 and 25-hydroxycholesterol stimulate osteoblast-like vascular cells to calcify. J Clin Invest 1994;93:2106-13.
192. Schober R, Hilbrich I, Jäger C, Holzer M. Senile plaque calcification of the lamina circumvoluta medullaris in Alzheimer's disease. Neuropathology 2021;41:366-70.
193. Hecht E, Freise C, Websky KV, et al. The matrix metalloproteinases 2 and 9 initiate uraemic vascular calcifications. Nephrol Dial Transplant 2016;31:789-97.
194. Zhang J, Wang J, Ma C, Lu J. Hydroxyapatite formation coexists with amyloid-like self-assembly of human amelogenin. Int J Mol Sci 2020;21:2946.
195. Obici L, Merlini G. An overview of drugs currently under investigation for the treatment of transthyretin-related hereditary amyloidosis. Expert Opin Investig Drugs 2014;23:1239-51.
196. Cox SJ, Lam B, Prasad A, et al. High-throughput screening at the membrane interface reveals inhibitors of amyloid-β. Biochemistry 2020;59:2249-58.
197. Tong XK, Hamel E. Simvastatin restored vascular reactivity, endothelial function and reduced string vessel pathology in a mouse model of cerebrovascular disease. J Cereb Blood Flow Metab 2015;35:512-20.
198. Roos CM, Zhang B, Palmer AK, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016;15:973-7.
199. Jing L, Li L, Ren X, et al. Role of sortilin and matrix vesicles in Nε-carboxymethyl-lysine-induced diabetic atherosclerotic calcification. Diabetes Metab Syndr Obes 2020;13:4141-51.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Whitehead M, Antonazzi M, Shanahan CM. Extracellular vesicles: the key to unlocking mechanisms of age-related vascular disease?. J Cardiovasc Aging 2024;4:12. http://dx.doi.org/10.20517/jca.2023.49
AMA Style
Whitehead M, Antonazzi M, Shanahan CM. Extracellular vesicles: the key to unlocking mechanisms of age-related vascular disease?. The Journal of Cardiovascular Aging. 2024; 4(2): 12. http://dx.doi.org/10.20517/jca.2023.49
Chicago/Turabian Style
Whitehead, Meredith, Marco Antonazzi, Catherine M. Shanahan. 2024. "Extracellular vesicles: the key to unlocking mechanisms of age-related vascular disease?" The Journal of Cardiovascular Aging. 4, no.2: 12. http://dx.doi.org/10.20517/jca.2023.49
ACS Style
Whitehead, M.; Antonazzi M.; Shanahan CM. Extracellular vesicles: the key to unlocking mechanisms of age-related vascular disease?. J. Cardiovasc. Aging. 2024, 4, 12. http://dx.doi.org/10.20517/jca.2023.49
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 3 clicks
Cite This Article 3 clicks

 Like This Article 6
likes
Like This Article 6
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.