
Molecular mechanisms underlying sarcopenia in heart failure
 0
0Abstract
The loss of skeletal muscle, also known as sarcopenia, is an aging-associated muscle disorder that is disproportionately present in heart failure (HF) patients. HF patients with sarcopenia have poor outcomes compared to the overall HF patient population. The prevalence of sarcopenia in HF is only expected to grow as the global population ages, and novel treatment strategies are needed to improve outcomes in this cohort. Multiple mechanistic pathways have emerged that may explain the increased prevalence of sarcopenia in the HF population, and a better understanding of these pathways may lead to the development of therapies to prevent muscle loss. This review article aims to explore the molecular mechanisms linking sarcopenia and HF, and to discuss treatment strategies aimed at addressing such molecular signals.
Keywords
INTRODUCTION
There are approximately 64 million patients suffering from heart failure (HF) globally[1], with 6.7 million cases situated within the United States[2]. Age is a major risk factor for the development of cardiovascular disease[3], and the prevalence of HF is known to increase with age[4]. Globally, the prevalence of HF continues to grow[5], and the total number of patients living with HF is expected to continue rising as the world population expands and ages[6]. Elderly patients, typically defined as those over 65 years old, represent the majority of HF cases, and the proportion of HF patients that are elderly is also expected to grow further in the coming years[7]. Age-associated diagnoses, including cachexia, frailty, and sarcopenia, complicate the diagnosis and management of HF, and are all associated with adverse HF-related outcomes[8,9].
Cachexia, frailty, and sarcopenia are all common age-associated clinical diagnoses with similar etiologies, characteristics, and diagnostic criteria[10-12]. There are nuanced clinical criteria underlying each of these diagnoses[9,12]. Broadly, frailty measures reduced functional capacity, cachexia represents weight loss, and sarcopenia refers to loss of peripheral muscle mass. The European Working Group on Sarcopenia in Older People (EWGSOP) has released guidance for the definition of sarcopenia as follows: patients with (1) low muscle strength have probable sarcopenia (as measured by grip strength or chair stand testing); while (2) low muscle quantity and quality confirms the diagnosis of sarcopenia (as measured by body imaging); and (3) low physical performance qualifies severe sarcopenia (as measured by performance testing; Table 1)[13]. This review will focus specifically on sarcopenia, which is associated with a number of adverse outcomes, including disability, poor quality of life, and death[13], and the molecular relationships between sarcopenia and HF.
European working group on sarcopenia in older people (EWGSOP2) clinical criteria for sarcopenia
| Measurement | Test and criteria according to EWGSOP2[13] |
| Muscle quantity | Grip strength (< 27 kg for men, < 16 kg for women) Chair stand test (> 15 s for five rises) |
| Muscle quality | Appendicular skeletal muscle mass (ASM; < 20 kg for men, < 15 kg for women) or ASM/height2 (< 7 kg/m2 for men, |
| Performance | Gait speed ≤ 0.8 m/s, ≤ 8 points on physical battery test, timed up and go test ≥ 20 s, or 400 m walk test ≥ 6 min or non-completion |
Pathologically, sarcopenia is characterized by loss of muscle fibers and accumulation of fat within muscle[14], and can be driven by immobility, nutritional deficit, inflammatory diseases, and genetic conditions, among other causes[15]. Sarcopenia is associated with multiple adverse outcomes, including impaired daily physical activities[16], increased incidence of falls and fractures[17], hospitalizations[18], and exacerbated mortality risk[19].
Sarcopenia is present between 20%-31% of HF patients[20,21], exceeding estimates of age-related muscle wasting alone[22]. Within the heart, myofibers have complex and varied changes during the development and progression of HF, including changes to myofibrillar protein composition and myofibrillar responses to neurohormonal signaling[23]. While HF may be associated with myofiber loss in some types of heart disease, such as dilated cardiomyopathy[24], myofibers are not necessarily lost in the heart in all HF patients. Loss of skeletal muscle myofibers, however, is a poor prognostic indicator in HF; its diagnosis is associated with poorer functional class and a higher incidence of cardiac events[25], while muscle wasting is a strong predictor of mortality in HF patients[26]. Sarcopenia has a similar prevalence in both HF patients with preserved ejection fraction (HFpEF) and reduced ejection fractions (HFrEF), while mortality in both HFpEF and HFrEF patients with sarcopenia is elevated at 1 year compared to non-sarcopenic patients[27].
Despite decades of research, resistance exercise remains the sole therapeutic strategy demonstrated to reliably improve measures of sarcopenia in the elderly population[28]. However, exercise tolerance is limited in HF patients, restricting the ability of patients to maintain prescribed exercise. Data on drug therapies designed to improve muscle mass and prevent body mass loss in advanced HF patients is sparse[29]. Traditional HF therapies exhibit a degree of beneficial effects in sarcopenic HF patients. For example, treatment with angiotensin-converting enzyme inhibitors (ACEi) was found to reduce the risk of weight loss in HF, though it did not improve measures of muscle mass[30]. In order to overcome this treatment deficit, a better understanding of the mechanistic signals underlying shifts in both cardiac and skeletal muscle within sarcopenic HF patients is required to facilitate the development of novel therapeutic strategies. This review will emphasize recent advances in the understanding of mechanisms linking HF and sarcopenia, and discuss potential therapeutic avenues for ameliorating these changes [Figure 1].
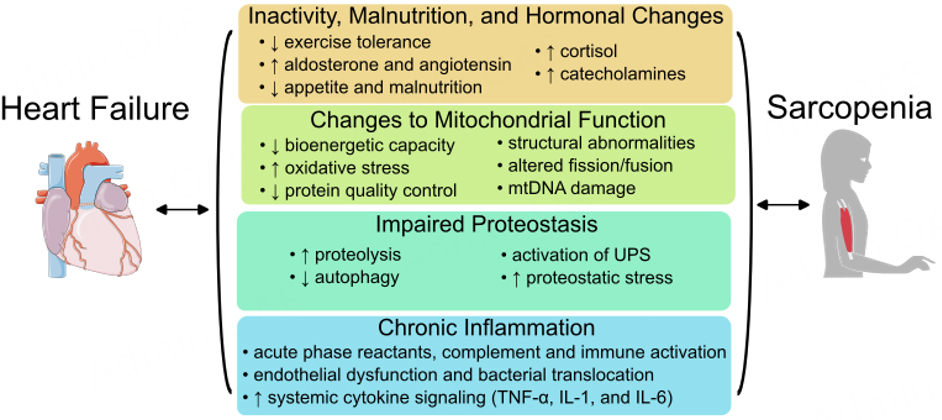
Figure 1. Molecular mechanisms linking heart failure (HF) and sarcopenia. HF patients have reduced exercise tolerance, malnutrition, and altered hormonal signaling, including changes to aldosterone, angiotensin, cortisol, and catecholamine levels, all of which are linked to muscle wasting. Both HF and sarcopenia are associated with mitochondrial changes, including loss of maximal energy production and altered fuel utilization, increased oxidative stress, changes to mitochondrial fission/fusion leading to structural abnormalities, mitochondrial DNA (mtDNA) damage, and reduced mitochondrial protein quality control. Both HF and sarcopenia also have altered protein production and turnover, most notably involving autophagy and the ubiquitin-protease system (UPS), leading to proteostatic stress. Finally, HF is associated with chronic inflammation, particularly as evidenced by increased cytokine signaling involving TNF-α,
INACTIVITY, MALNUTRITION, AND HORMONAL CHANGES
Perhaps the most intuitive link between HF and sarcopenia involves physical inactivity in the HF patient. Exercise intolerance and reduced functional capacity are both hallmarks of HF[31] and risk factors for developing sarcopenia[22]. Exercise training, particularly resistance exercise, remains the most well-studied and reliable method for improving muscle function in HF patients[32,33], despite the difficulties of implementing exercise training in the HF population[31]. However, the hormonal, nutritional, and metabolic changes linking HF and sarcopenia are much more complex than solely attributed to lack of physical activity [Figure 2].

Figure 2. Overview of activity-related, nutritional, and hormonal changes in heart failure (HF) leading to muscle loss. HF patients have exercise intolerance and reduced mobility, leading to reduced exercise capacity. Some HF patients have reduced appetite or digestive issues in the gastrointestinal (GI) tract, leading to malnutrition and reduced protein intake, limiting the ability to sustain muscle growth. HF is also associated with increased renin-angiotensin-aldosterone system (RAAS) signaling, which promotes muscle loss over time. Finally, hormonal changes seen in HF, including growth hormone, catecholamine, ghrelin, and testosterone signaling, all drive an imbalance in catabolism over anabolism, resulting in muscle loss.
Patients with cachexia in HF are known to have dysregulated neurohormonal signaling compared to non-cachectic patients, including shifts to circulating levels of aldosterone, norepinephrine, epinephrine, cortisol, tumor necrosis factor (TNF), and human growth hormone (GH)[34]. Broadly, these shifts are associated with an increase in catabolism and energy expenditure, signaling a mismatch between anabolic and catabolic processes that may underly proteostatic changes in the muscle of HF patients who are losing weight. While targeting such a broad milieu of hormonal shifts is pharmaceutically challenging, one obvious strategy to tip this imbalance towards a healthy compromise is to optimize nutrition in order to meet the increased catabolic needs of the advanced HF patient.
Loss of appetite affects a significant portion of HF patients (10%) and is associated with worse one-year mortality in patients hospitalized with acute, decompensated HF[35]. Appetite stimulants, such as megestrol and dronabinol, have been investigated for elderly patients with failure to thrive in the general population, and are typically not recommended[36]. Cachectic cancer patients, who have significant overlap in disease presentation and pathophysiology to cachectic heart failure patients[37], are occasionally prescribed appetite stimulants to counteract weight loss. While these drugs have been found to improve body weight in this population, though not necessarily muscle mass, they do not improve mortality[38]. There is less evidence available for appetite stimulants in HF patients with weight and muscle loss. While megestrol was found to improve lean muscle mass and cardiac function in a rat model of cachexia-induced cardiomyopathy[39], no human data exists to support its use in HF patients with muscle loss. A wide number of studies have focused on micronutrient supplementation and overall HF outcomes[40], though only a few studies focused on muscle mass as an endpoint. Nutritional interventions, particularly increased protein intake, may offer benefits for HF patients with sarcopenia, though there is no broad clinical data to support improved outcomes related to increased protein consumption[41]. However, general best-practice recommendations typically involve a nutrient-rich diet with anti-inflammatory properties, such as polyunsaturated fatty acids, and a Mediterranean-style diet, with hypercaloric or hyperproteic supplements if oral nutrition is not effective[40]. Currently, studies involving Mediterranean, hypercaloric, and hyperproteic diets in HF patients with sarcopenia are underway[42]. Increased protein and amino acid intake could be simple interventions for patients with muscle loss, and could be of particular importance in HF patients with sarcopenia, though larger-scale human studies are required to support this.
Beyond nutritional support, efforts have been made to evaluate specific hormonal targets to improve muscle mass and function in the HF population. Angiotensin signaling, a well-known hormonal change in HF, has been linked to elevated skeletal muscle catabolism through multiple mechanisms, including indirect effects such as vasoconstriction of blood vessels supporting muscle and promotion of mitochondrial dysregulation [Figure 3][43], as well as direct effects like activation of the ubiquitin-proteasome proteolytic pathway
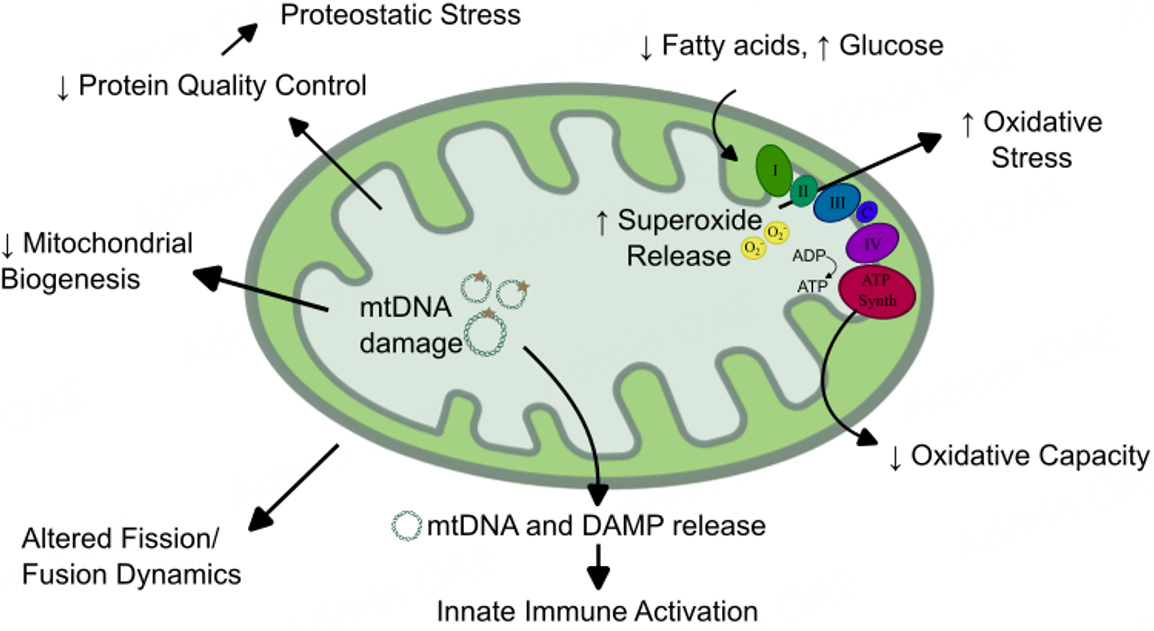
Figure 3. Changes to mitochondrial activity, behavior, and function common to failing myocardium and muscle tissue in sarcopenia. Mitochondria carry out a wide breadth of essential functions in the heart and muscle, and nearly all of these functions are dysregulated in heart failure and sarcopenia. Changes along the electron transport chain (Complexes I-IV and ATP synthase) lead to reduced ATP generation and increase superoxide release. Fuel utilization shifts from predominately fatty acids to alternative fuels, including glucose. Mitochondrial DNAs (mtDNAs) are damaged and released, leading to reduced mitochondrial biogenesis and activation of the innate immune response. Similarly, mitochondrial damage-associated molecular patterns (DAMPs) are exposed, triggering an immune response. Mitochondrial dynamics, including fission and fusion of established mitochondria, are altered, and reduced protein quality control can cause proteostatic stress in the cell.
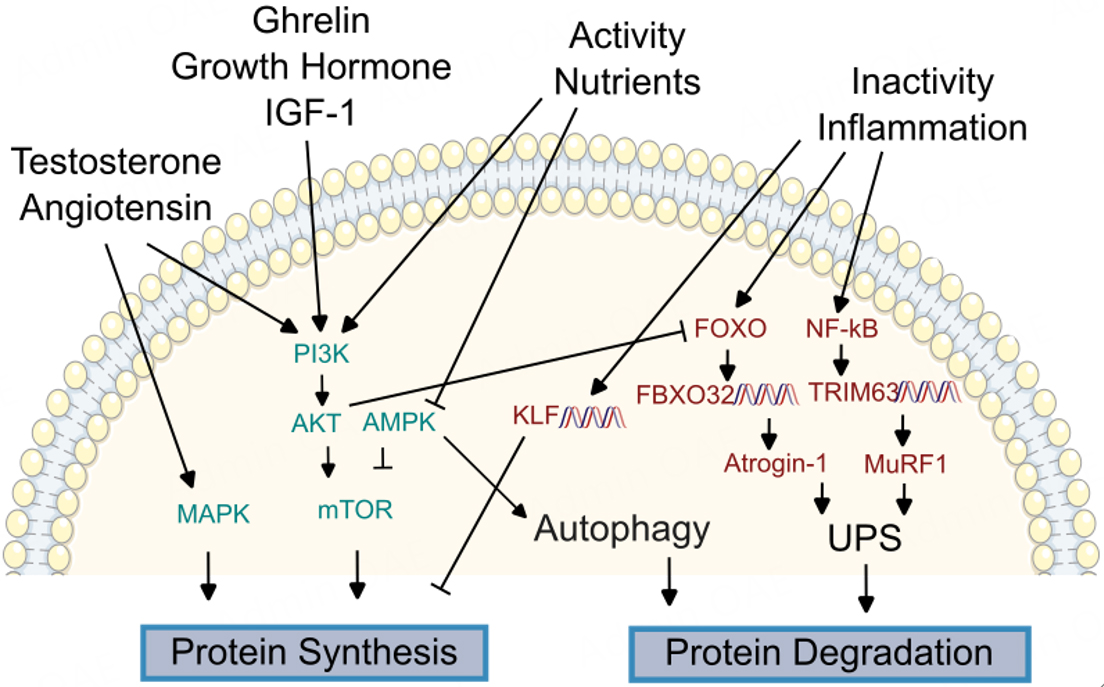
Figure 4. Overview of molecular regulators of proteostasis. Maintenance of the proteome, including synthesis of new proteins and degradation of old and damaged proteins, is dysregulated in both the failing heart and sarcopenic muscle. Signal transducers linking hormonal and nutritional changes to protein synthesis are conserved between the heart and peripheral muscle, including mitogen-activated protein kinase (MAPK), phosphatidylinositol-3-kinase (PI3K) / protein kinase B (AKT), AMP-activated protein kinase, and mammalian target of rapamycin (mTOR). Signals promoting proteolysis include Forkhead box O (FoxO) proteins, which can promote transcription of F-box protein 32 (FBXO32) to increase expression of Atrogin-1 to activate the ubiquitin protease system (UPS). Similarly, activation of nuclear factor κB (NF-κB) triggers transcription of TRIM63 to drive Muscle RING-finger protein-1 (MuRF-1) expression and promote UPS. Kruppel-like factors (KLFs) are promoted by inactivity to inhibit protein synthesis.
Ghrelin, a peptide generated in the gastrointestinal tract that promotes appetite and alters energy metabolism[47,48], could be a more feasible target than broader appetite stimulants. Circulating ghrelin levels are dysregulated within cachectic HF patients compared to HF patients without cachexia[49]. Ghrelin analogs reduce myostatin expression, an inhibitor of muscle growth, in an animal model of HF post-myocardial infarction, providing a direct mechanistic link between ghrelin and muscle loss[50]. Among its many other activities, circulating ghrelin promotes cortisol and growth hormone (GH) secretion, and also stimulates insulin-like growth factor (IGF)-1, which promotes protein synthesis via the phosphatidylinositol-3-kinase (PI3K)/protein kinase B (PKB or AKT) pathway (see proteostasis section below for further details;
Another promising hormonal target for altering age-related muscle disorders in HF patients is testosterone. Short-term testosterone supplementation has demonstrated improved outcomes in walk-tests in HF patients[55-57], as well as increased muscle mass[58] and improved hand grip[59], albeit with less clear benefits as treatment extended to one year[60,61]. While there has been historical concern concerning testosterone supplementation increasing major cardiovascular effects, recent data supports testosterone’s safety in elderly men with hypogonadism[62]. The exact mechanism of testosterone on muscle mass remains unclear. In vitro studies have found that testosterone increases IGF-1 expression, which may promote protein synthesis via PI3K/AKT signaling, as seen with ghrelin supplementation[63]. The increase in protein synthesis has been confirmed in human studies[64]. Other in vitro studies have linked testosterone supplementation to increased mitotic activity in myoblast cells[65], while another found that testosterone’s effects depend on the mammalian target of rapamycin (mTOR), a protein kinase that plays a major role in cell growth and metabolism[66] [Figure 3]. Regardless of its mechanism, inconsistent clinical benefits and a history of reported adverse cardiovascular events remain major barriers to testosterone’s use for the prevention of sarcopenia in HF.
Recently, novel diabetic therapies have come to market with substantial utilization in patients having cardiovascular disorders[67]. Specifically, sodium-glucose cotransporter-2 inhibitors (SGLT2i) and glucagon-like peptide-1 receptor agonists (GLP-1RA) have rapidly increased in popularity within the United States. Their effects on the prevalence of sarcopenia in HF have not yet been fully evaluated, though early reports suggest that both classes of drugs could play a role in muscle loss. SGLT2i drugs inhibit glucose reabsorption within renal proximal tubules to promote urinary glucose excretion. These drugs have recently been recommended for the management of HF[68]. SGLT2i were associated with reduced muscle mass in some, though not all, clinical trials in diabetic patients[69]. The mechanism of skeletal muscle shifts and cardiovascular benefits of SGLT2i drugs remain unclear, particularly as the primary effect of any SGLT2i appears to occur renally, and a broad number of hypotheses, including fatty acid and ketone metabolism, mitochondrial shifts, nitric oxide availability, and improved endothelial function, are being explored[70,71]. However, most current HF studies of SGLT2i are focused on cardiac musculature, rather than peripheral muscle changes. Given the rapid utilization of SGLT2i in HF cohorts, a better understanding of its effects on skeletal muscle in HF patients is warranted.
Similarly, GLP-1RAs, another class of diabetic medications with cardiovascular benefits, may affect skeletal muscle mass and function[72]. GLP-1RAs stimulate insulin production and delay gastric emptying, resulting in improved glucose regulation and increased satiety, leading to benefits in the management of both diabetes and obesity[73]. Though GLP-1RA’s benefits in HF have been predominantly demonstrated in preclinical models, their mass adoption as weight-loss agents will likely affect HF and sarcopenia outcomes on a population level[74]. GLP-1RAs have had mixed reports on muscle mass in animal models, as beneficial effects attributed to anti-inflammatory properties[75] and suppression of myostatin[76] have been identified in aging models. Angiotensin-challenged mice demonstrated reduced muscle mass when treated with a
MITOCHONDRIAL ACTIVITY, BEHAVIOR, AND FUNCTION
Mitochondrial dysfunction is an overly broad term, as mitochondria serve a wide breadth of functions, including, but not limited to, energy production, ion regulation, redox homeostasis, regulation of apoptosis, inflammatory signaling, and steroidogenesis[77]. Nearly all of these mitochondrial roles are reduced or dysregulated in the myocardium during HF[78]. Mitochondria are established central figures in the pathogenesis of HF, and recent transcriptomic and metabolomic studies in HF models have confirmed their importance[79]. Mitochondrial shifts are also heavily implicated in normal aging. Dysregulated mitochondrial fuel metabolism, increased oxidative stress, mitochondrial DNA damage, loss of mitochondrial protein quality control, and altered mitochondrial fission and fusion are all hallmarks of the aging heart[80].
Much like the myocardium, skeletal muscle is highly oxidative and typically contains high mitochondrial density, though mitochondrial content varies significantly by muscle type[81]. Skeletal muscle from models of aging-associated muscle disorders demonstrates comparable mitochondrial shifts to those seen in both the aging and failing heart, all of which are generally associated with reduced mitochondrial performance or deleterious mitochondrial signaling to other organelles. These changes include reduced total mitochondrial content[82], mitochondrial structural remodeling[83], altered energetics[84] and reduced maximal bioenergetic capacity[85], increased mitochondrial oxidative stress and dysregulated redox signaling[86,87], reduced mitochondrial protein quality control[80], and increased mitochondria-associated inflammation[84,88]. These broad similarities to mitochondrial changes between the failing heart and aging skeletal muscle suggest significant overlap in mitochondrial pathophysiology.
Among the many mitochondrial functions necessary in both the myocardium and skeletal muscle, maintenance of energy through ATP generation has received the most intensive research focus. Altered mitochondrial fuel use has long been known to be a major issue underlying HF, as the failing heart has been described as “an engine out of fuel”[89] and is one of the earlier detectable mitochondrial shifts in the failing heart. Skeletal muscle bioenergetics, including mitochondrial ATP production and substrate utilization, as well as mitochondrial biogenesis, are all decreased in HF patients[90]. HF patients also have lower rates of oxidative phosphorylation and longer recovery times in skeletal tissue by nuclear magnetic resonance spectroscopy[91], as well as decreased mitochondrial fatty acid oxidation and increased glucose utilization. Such manifestations are detectable early in cardiac disease, occurring even prior to decreased cardiac contractility[92]. Similarly, dysregulated mitochondrial respiration is one of the earliest shifts in skeletal muscle, as demonstrated in HF rodents[93]. Skeletal muscle shifts in HF animals follow a similar trajectory to cardiac muscle when measuring mitochondrial respiratory capacity in a HF model[94], again supporting a common mitochondrial phenotypic change between myocardium and skeletal muscle. Though these changes are not commonly measured clinical parameters, advances in respirometry protocols and utilization of circulating platelets as tissue alternatives for evaluating mitochondrial respiratory capacity[95] may eventually lead to clinical testing of bioenergetic capacity with diagnostic and prognostic value.
Peripheral muscle in HF patients also has increased reactive oxygen species (ROS) and mitochondrial structural abnormalities[96], though it is not clear if these are the consequences or drivers of mitochondrial bioenergetic shifts. Mitochondria are the major source of ROS in the heart[97] and a major contributor to ROS generation in peripheral muscle[98]. Incomplete reduction of oxygen along the electron transport chain (ETC) can produce superoxide, which can cause broad oxidative damage to the cell, and further mitochondrial damage locally, including damage to mitochondrial DNA (mtDNA).
Mitochondria contain a number of damage-associated molecular patterns (DAMPs) that may trigger the innate immune response[99], as discussed later in this review. In particular, mtDNA, which is highly similar to bacterial DNA, can drive immune activation when released from the mitochondria[100]. The most prominent inflammatory cascade related to mtDNA involves activation of the cyclic GMP-AMP synthases (cGAS) pathway, which activates interferon signaling via the Stimulator of Interferon Genes (STING) protein[101,102], a pathway known to increase with aging[100]. mtDNAs also lack many of the complex repair enzymes present in nuclear DNA, so they are highly prone to oxidative stress and damage[103]. mtDNA damage has been demonstrated extensively in heart disease[104,105] and sarcopenia[106], which in turn lead to ETC dysfunction and reduced mitochondrial biogenesis[105].
The role of mitochondria as either a primary cause or consequence of other signaling pathways common to cardiac and skeletal muscle remains unclear. Systemic signals prevalent in HF, such as increased adrenergic stimulation and renin-angiotensin signaling, are crucial upstream regulators leading to dysregulated mitochondrial bioenergetics in both myocardium and skeletal muscle[90,107], and are already targeted as pillars of HF treatments. Such therapies, including drugs targeting adrenergic and renin-angiotensin cascades such as β-blockers and ACEis, respectively, are known to have myriad cardiac mitochondrial benefits[108]. ACEis were found to counter deleterious mitochondrial shifts in the peripheral muscle of rats with HF post-myocardial infarction, leading to improved musculature-based oxidative capacity and normalized expression of mitochondrial transcription factors[109], though the effects of β-adrenergic blockade directly on mitochondria are less clear[110]. More broadly, endurance exercise regimens, which remain the strongest recommendation for improving sarcopenia in HF, are known to improve mitochondrial morphology and dynamics in the skeletal muscle of aging patients[111].
Despite the evidence of mitochondrial involvement and broad shifts to mitochondrial functions within both cardiac and skeletal muscle in HF patients, therapies directly targeting the mitochondria remain underdeveloped. Exercise and caloric restriction, which have been shown to protect against mitochondrial disease associated with aging animals, are limited in the HF population[112,113]. Mitochondrial ROS scavengers have demonstrated multiple benefits in preclinical models of cardiac disease, though these therapies have largely failed to translate into clinical benefits in human trials[114]. Strategies aimed at restoring the balance of nicotinamide adenine dinucleotide (NAD+) and its reduced form (NADH), which are coenzymes that are responsible for carrying electrons and crucial regulators of redox metabolism in the mitochondria[115], could be a more viable mitochondrially-targeted therapy. Supplementation with NAD+ and treatment with nicotinamide riboside, a precursor to NAD+, works to remedy the depleted NAD+ pool that has been causally linked to myocardial mitochondria in HF[116]. Restoring NAD+ levels was also found to be beneficial in models of aging muscle[117]. Targeting NAD+ could be a promising and easily translatable approach to improving mitochondrial bioenergetic functions in both the myocardium and skeletal muscle of HF patients. An extensive review of mitochondria-targeted therapies for skeletal muscle disorders in HF patients was recently published by Lv et al.[88].
Given the breadth of mitochondrial changes seen in failing myocardium and sarcopenic muscle, as well as significant overlap in mitochondrial pathophysiology between these tissues, efforts to ameliorate mitochondrial changes and develop mitochondrially targeted therapies should be promoted in sarcopenic HF models. Work investigating mitochondria in such models should focus on specific mitochondrial activities or functions, be they energetic, signaling, ROS, inflammatory, or proteostatic changes, rather than broadly labeling mitochondrial dysfunction as a driver of disease.
PROTEOSTASIS
Healthy muscle, in both the heart and peripherally, requires constant synthesis of new proteins together with degradation of old and damaged components, a process known collectively as proteostasis. Proteostasis relies on a number of mechanisms to maintain the proteome, including regulation of protein machinery such as ribosomes, utilization of chaperone proteins to promote protein folding, degradation pathways, such as the ubiquitin-proteasome system (UPS), and autophagy by lysosomes, as well as feedback mechanisms to identify misfolded proteins and protein accumulation[118] [Figure 4]. Dysfunction of proteostasis is a hallmark of aging[119], triggering a stress response in cells. If left unchecked, this dysfunction may eventually overwhelm the cell, leading to cell death or systemic stress signaling via the unfolded protein response[120]. Cardiomyocytes are terminally differentiated, so the removal of protein aggregates and damaged protein is critical to avoiding cell death and cardiac fibrosis. Alterations in cardiac proteostasis have been demonstrated within several cardiac disease models, including ischemia[121], arrhythmia[122], and heart failure[123], as well as in the aging heart[124]. Alterations in muscle proteostasis are critical changes in the development of sarcopenia as well[125].
There is significant overlap in the molecular regulators of protein anabolism and catabolism between the heart and peripheral muscle. These include highly conserved metabolic sensors, such as PI3K/AKT, AMP-activated protein kinase (AMPK), and mitogen-activated protein kinase (MAPK). As discussed earlier in this review, PI3K/AKT is a central signaling cascade linking external stimuli, such as insulin, GH, or growth factors, to transcriptional regulators of cell growth and protein synthesis[126]. Similarly, AMPK is a critical regulator of energy homeostasis and a metabolic sensor[127]. AMPK is activated when ATP is depleted, as occurs in cardiac and skeletal muscle tissue in ischemia[121,128], nutrient deprivation[129], and exercise[130]. Activated AMPK promotes a wide number of cellular processes, including mitochondrial biogenesis, inflammation, and autophagy, while inhibiting energy intensive processes, such as protein synthesis[131]. AKT and AMPK can interact with the activity of each other, and also share a common downstream effector, the mammalian target of rapamycin (mTOR)[66], another protein kinase that plays a major role in cell growth and metabolism[66]. Therapies affecting mTOR activity, such as rapamycin, metformin, and resveratrol, are of high interest as anti-aging therapies[132]. Finally, MAPK is a highly conserved kinase family able to transduce extracellular changes, including exercise, cytokine signals, and cell stress[133], to regulators of cell proliferation and survival[134]. MAPK and AKT have both been identified as downstream regulators for both testosterone[135] and angiotensin[136].
Given the breadth of functions and tissue ubiquity of AKT, AMPK, and MAPK signaling cascades, targeting more specific molecular signals may offer improved insight and therapeutic opportunities towards the prevention of sarcopenia. Regulators of proteolysis may offer such specificity. Aged muscle tissue has increased biomarkers of proteolysis than younger cohorts. While proteasomal activity does not necessarily decrease with age, as measured in muscle biopsies from young versus aged healthy adults[137,138], other biomarkers of proteostatic dysfunction, including impaired autophagy and endoplasmic reticulum stress, have been well-reported in sarcopenia[125]. Molecular markers of proteolysis are conserved between heart and muscle in models of skeletal muscle atrophy and heart failure. Such conserved markers include Muscle RING-finger protein-1 (MuRF-1), an E3 ubiquitin ligase that plays a crucial role in protein ubiquitinylation and subsequent degradation[139], and F-box protein 32 (FBXO32, also known as MAFBX or atrogin 1), another E3 ubiquitin ligase.
MuRF-1 is expressed in cardiac and skeletal muscle and is encoded by the gene TRIM63[140]. MuRF-1 is upregulated in cell models of skeletal muscle atrophy[141] and in muscle tissues of sarcopenic rats[142]. In the heart, MuRF-1 inhibits cardiac hypertrophy in a surgical mouse model of heart failure[143] and has been proposed as a therapy for cardiac hypertrophy in mice[144], suggesting that MuRF-1 has common effects on myofibers across tissues. Overexpression of MuRF-1 can also result in thinning of cardiac muscle and cardiac dysfunction[145]. HF can also cause changes to MuRF-1 and proteasomal activity in peripheral muscle. In a rat model of HF, protein expression of MuRF-1 and proteasomal activity is increased in both the diaphragm and quadriceps mucles[146]. In humans, MuRF-1 is increased in the skeletal muscle of HF patients, and can be blunted by exercise[147]. This may explain, at least in part, the beneficial effects of exercise in HF patients, such as increased protein synthesis and decreased protein degradation that have been demonstrated with exercise in a HF cohort[148]. FBXO32 signaling correlates with MuRF-1 in the heart and skeletal muscle, and FBXO32 is highly expressed in muscle tissue during muscle atrophy[149,150], upregulated in animal models of heart failure[151], and down-regulated in cardiac atrophy[152]. Similar to MuRF-1, FBXO32 mRNA levels increase in the skeletal muscle of HF rats[153].
The failing myocardium and sarcopenic muscle share transcriptional signals related to proteostasis beyond markers of proteolysis. Two examples are Kruppel-like factors (KLFs), which are DNA-binding proteins that partially control genes for cell growth and differentiation[154], and Forkhead Box O genes (FOXO)[155], which are discussed further in the inflammation section. KLF15 is a major pathway involved in immobilization-induced skeletal muscle atrophy in mice[156] and a wide family of KLF genes have been implicated in heart disease, including HF[154]. KLFs have myriad roles, including regulating fuel utilization in both skeletal muscle and the heart[157,158], supporting mitochondrial biogenesis in the heart[159], and mediating vascular inflammatory changes[160].
Autophagy is another important mediator of healthy proteostasis shared between peripheral muscle and the heart. Autophagy is induced by exercise training to promote the healthy turnover of muscle tissue[161,162]. However, nutrient deprivation can trigger autophagy through AMPK activation, leading to excessive muscle loss and muscle atrophy[163]. Mutations causing excessive autophagy can lead to disproportionate muscle loss in transgenic mice[164]. Shifts to autophagy occur early in the progression of muscle loss. Increased markers of autophagy, as well as activation of the UPS, can be detected early within the peripheral muscle of HF rats, prior to evidence of muscle loss that can be detected physiologically[165]. Similarly, the upregulation of autophagy-associated genes occurs early after infarction in the peripheral muscle of rats in a myocardial infarction model[166]. If unchecked, these shifts may lead to apoptosis in skeletal muscle, which is known to occur in HF patients[167], leading to worsened muscular outcomes. While the search for therapies to manipulate UPS[168] and autophagy in sarcopenia is underway[169], any potential therapies will need to find a healthy balance to maintain muscle, while also promoting alternative pathways of muscle destruction.
INFLAMMATION
Immune activation in HF is widely reported[170,171], with evidence of elevated serum inflammatory markers, specifically tumor-necrosis factor-alpha (TNF-α), in chronic HF patients dating back over 30 years[172]. Since that initial observation, a broad number of inflammatory markers, including acute phase reactants, cytokines, and activation of the complement system, have been linked to HF patients[173], both intrinsic and extrinsic to the heart[170,173]. Dysregulated circulating cytokine levels can be detected within the blood of HF patients, and several cytokines are predictive of mortality in these cohorts[174,175]. Normal human aging has also been linked to dysregulated markers of chronic inflammation, particularly in cardiac stress[176]. While the wide breadth of immune activation and various biomarkers of chronic inflammation in HF is beyond the scope of this review, a number of cytokine signals have been linked more directly to muscle wasting in HF patients. Most notably, these include cytokines TNF-α, interleukin (IL)-1, and IL-6 [Figure 5].

Figure 5. Overview of cytokine changes in myocardium and skeletal muscle during Heart Failure (HF). HF can drive a wide breadth of inflammatory changes. Some of these include elevated pro-inflammatory hormonal signaling, gut edema allowing bacterial translocation, and exposure of damage-associate molecular patterns (DAMPs), leading to the release of cytokines including tumor necrosis factor (TNF) and interleukins (IL) 1 and 6. TNF can cause apoptosis, elevated reactive oxygen species (ROS), and mitochondrial DNA (mtDNA) damage in the musculature, leading to muscle damage and degradation. Transcription factors, including the Forkhead box O (FOXO) nuclear factor κB (NF-κB) families, are activated by cytokines, leading to altered proteostasis and activation of the ubiquitin-protease system (UPS).
TNF-α has been directly linked to the development of HF in animal models. Administration of systemic TNF-α was found to cause the development of LV dysfunction in a rat model[177]. Cardiac-specific TNF-α overexpression in transgenic mice not only causes LV dysfunction, but also leads to the disruption of muscle function, including the diaphragm[178]. HF patients with elevated levels of circulating TNF have more advanced HF in comparison to HF patients with reduced TNF levels[172]. Higher levels of TNF-α, as well as IL-6 receptor levels, are associated with loss of grip strength and muscle mass in an aging population[179], and serum TNF-α levels are a strong predictor of weight loss in HF patients[34].
Mechanistically, Excess TNF-α signaling can promote catabolism through a number of downstream pathways. TNF-α promotes caspase-mediated apoptosis in skeletal muscle, particularly in type II fibers, which may help explain shifts in muscle fiber composition in the aging population[180]. TNF-α activation is also linked to dysregulated proteostasis, as TNF-α-treated cachectic rats have reduced protein turnover and increased weight loss compared to untreated controls[181]. TNF-α also causes a number of deleterious shifts in cardiac and peripheral muscle tissue, and is known to promote ROS[182] and damage mitochondrial DNA[183].
Similar to TNF-α, the pro-inflammatory cytokines IL-1 and IL-6 are also associated with weight loss, muscle wasting, and frailty in the general population[184], and both have been linked to worsened HF functional class[185]. Both IL-1 and Il-6 can trigger activation of nuclear factor κB (NF-κB), a broad family of transcription factors that helps regulate the cell response to infection and can trigger both the innate and adaptive immune response[186] and activation of proteolysis-associated genes[187]. NF-κB transcription in muscle cells can lead to activation of the UPS and muscle degradation[32]. IL-6 signaling has long been associated with the development of cachexia in cancer patients[188] and, more recently, has been causally linked to muscle wasting in mice[189]. In HF patients, systemic IL-6 levels are higher in patient cohorts with muscle wasting compared to HF patients without muscle loss[21]. Data on IL-1 association with weight loss in HF patients, however, has been mixed[21,190]. IL-1 has a diverse role in functions involving both myocardial contractility and regulation of food intake in the hypothalamus, rendering it a clear candidate to link HF and weight loss[191], although its direct link to muscle loss in HF patients has not yet been proven.
Similar to NF-κB, The Forkhead box O (FoxO) proteins are another family of transcription factors known to be dysregulated in both heart failure[192] and sarcopenia[155]. FoxO proteins can be activated by oxidative stress, nutrient deprivation, or DNA damage and regulated by PI3K-Akt signaling[192]. More recently, FoxO family signaling has been linked to cytokine signaling in skeletal muscle[193], leading to increased atrogin-1 and promoting muscle catabolism.
While cytokine signaling is one of the best-known links between diseases of aging and muscle loss, in some part due to the ease with which cytokine profiles can be measured in circulating blood, a number of tissue intrinsic pathways also promote immune response. One such pathway involves DAMPS, particularly from the mitochondria. Release of ATP, cardiolipin, and mtDNA from the mitochondria can trigger immune activators including the cGAS-STING pathway, driving pro-inflammatory interferon release[194], or activating the nucleotide oligomerization domain-, leucine-rich repeat-, and pyrin domain-containing 3 protein (NLRP3) inflammasome, which can trigger caspase activity and further drive IL-1 cytokine release[195].
One physiologic mechanism linking HF to immune activation involves congestion of mesenteric veins secondary to fluid overload in the HF patient. This congestion allows for bacterial translocation and endotoxin release from the gut. This concept was originally proposed by Anker et al. and has expanded over the last 25 years[196], as has recently been summarized in a review by Liu et al.[197]. Endothelial cell dysfunction and vessel contractility have long been linked to HF[198], and HF patients with sarcopenia are known to have reduced peripheral blood flow and impaired endothelial cell function compared to HF patients without sarcopenia[199]. Reduced peripheral blood flow reduces exercise capacity in HF patients and limits the nutritional support required to sustain peripheral musculature[200]. Endothelial cells are major players in the inflammatory process, having varied responses to acute and chronic inflammation that could lead to hyperemia, dysregulated permeability, angiogenesis, and dysregulated blood flow[201]. Within the intestinal tract, these alterations could lead to intestinal edema and bacterial translocation, leading to potentially dramatic shifts to systemic inflammation. This mechanism links the fluid overload, common to HF patients, to immune activation via the gastrointestinal tract, and supports a link between diuretic therapy and reduced inflammation in the HF population.
Unfortunately, systemic anti-cytokine therapies have not been found to improve mortality or hospitalization rates in HF patients. Though specific outcomes related to muscle mass have not been reported, treatment with etanercept, a TNF inhibitor, did not improve clinical status in HF patients[202]. Similarly, treatment with infliximab, a monoclonal antibody to TNF-α, did not improve HF outcomes[203]. Importantly, neither of these focused specifically on biomarkers of sarcopenia. The field of targeted anti-inflammatory therapies is rapidly growing[204], and novel anti-cytokine and anti-inflammatory compounds could offer beneficial effects towards HF-associated sarcopenia in the future.
CONCLUSIONS
Age-associated muscle disorders, particularly sarcopenia, are associated with advanced HF and lead to poor outcomes and high mortality rates. The prevalence of muscle wasting in HF is expected to grow as HF patients live longer and the global population continues to age. However, the mass adoption of novel drug therapies, including SGLT2is and GLP-1Ras, could shift the prevalence of HF sarcopenia in unpredictable ways. There is a myriad of proposed mechanisms linking HF to peripheral muscle wasting, including limited physical activity and malnutrition in HF patients, abnormal mitochondrial function, dysregulated proteostatic signaling, and systemic inflammation, all of which could be contributing to muscle loss. While traditional HF therapies, including ACEi, β-blockers, and resistance exercise, remain the strongest recommendations for preventing muscle loss, there is promising data supporting testosterone, ghrelin, and mitochondrial-targeted therapies that could gain prominence, though continued human trials and increased knowledge of molecular mechanisms are still critically needed.
DECLARATIONS
Author’s Contributions
The author contributed solely to the article.
Availability of data and materials
Not applicable.
Financial support and sponsorship
Dr. Cody A. Rutledge is supported by grants from the Veterans Health Administration (IK2BX005785).
Conflicts of interest
The author declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2024.
REFERENCES
1. Becher PM, Lund LH, Coats AJS, Savarese G. An update on global epidemiology in heart failure. Eur Heart J 2022;43:3005-7.
2. Tsao CW, Aday AW, Almarzooq ZI, et al. Heart disease and stroke statistics-2023 update: a report from the american heart association. Circulation 2023;147:e93-621.
3. Li H, Hastings MH, Rhee J, Trager LE, Roh JD, Rosenzweig A. Targeting age-related pathways in heart failure. Circ Res 2020;126:533-51.
4. Ho KK, Pinsky JL, Kannel WB, Levy D. The epidemiology of heart failure: the Framingham study. J Am Coll Cardiol 1993;22:6A-13A.
5. Savarese G, Becher PM, Lund LH, Seferovic P, Rosano GMC, Coats AJS. Global burden of heart failure: a comprehensive and updated review of epidemiology. Cardiovasc Res 2023;118:3272-87.
6. Groenewegen A, Rutten FH, Mosterd A, Hoes AW. Epidemiology of heart failure. Eur J Heart Fail 2020;22:1342-56.
8. Uchmanowicz I, Gobbens RJ. The relationship between frailty, anxiety and depression, and health-related quality of life in elderly patients with heart failure. Clin Interv Aging 2015;10:1595-600.
9. Beltrami M, Fumagalli C, Milli M. Frailty, sarcopenia and cachexia in heart failure patients: different clinical entities of the same painting. World J Cardiol 2021;13:1-10.
10. Jeejeebhoy KN. Malnutrition, fatigue, frailty, vulnerability, sarcopenia and cachexia: overlap of clinical features. Curr Opin Clin Nutr Metab Care 2012;15:213-9.
11. Petermann-Rocha F, Pell JP, Celis-Morales C, Ho FK. Frailty, sarcopenia, cachexia and malnutrition as comorbid conditions and their associations with mortality: a prospective study from UK biobank. J Public Health 2022;44:e172-80.
12. Mirzai S, Eck BL, Chen PH, Estep JD, Tang WHW. Current approach to the diagnosis of sarcopenia in heart failure: a narrative review on the role of clinical and imaging assessments. Circ Heart Fail 2022;15:e009322.
13. Cruz-Jentoft AJ, Bahat G, Bauer J, et al. Sarcopenia: revised European consensus on definition and diagnosis. Age Ageing 2019;48:16-31.
14. Muscaritoli M, Anker SD, Argilés J, et al. Consensus definition of sarcopenia, cachexia and pre-cachexia: joint document elaborated by Special Interest Groups (SIG) “cachexia-anorexia in chronic wasting diseases” and “nutrition in geriatrics”. Clin Nutr 2010;29:154-9.
15. Ali S, Garcia JM. Sarcopenia, cachexia and aging: diagnosis, mechanisms and therapeutic options - a mini-review. Gerontology 2014;60:294-305.
16. Wang DXM, Yao J, Zirek Y, Reijnierse EM, Maier AB. Muscle mass, strength, and physical performance predicting activities of daily living: a meta-analysis. J Cachexia Sarcopenia Muscle 2020;11:3-25.
17. Yeung SSY, Reijnierse EM, Pham VK, et al. Sarcopenia and its association with falls and fractures in older adults: a systematic review and meta-analysis. J Cachexia Sarcopenia Muscle 2019;10:485-500.
18. Beaudart C, Zaaria M, Pasleau F, Reginster JY, Bruyère O. Health outcomes of sarcopenia: a systematic review and meta-analysis. PLoS One 2017;12:e0169548.
19. Xu J, Wan CS, Ktoris K, Reijnierse EM, Maier AB. Sarcopenia is associated with mortality in adults: a systematic review and meta-analysis. Gerontology 2022;68:361-76.
20. Chen R, Xu J, Wang Y, et al. Prevalence of sarcopenia and its association with clinical outcomes in heart failure: an updated meta-analysis and systematic review. Clin Cardiol 2023;46:260-8.
21. Fülster S, Tacke M, Sandek A, et al. Muscle wasting in patients with chronic heart failure: results from the studies investigating co-morbidities aggravating heart failure (SICA-HF). Eur Heart J 2013;34:512-9.
22. Yuan S, Larsson SC. Epidemiology of sarcopenia: prevalence, risk factors, and consequences. Metabolism 2023;144:155533.
23. Machackova J, Barta J, Dhalla NS. Myofibrillar remodeling in cardiac hypertrophy, heart failure and cardiomyopathies. Can J Cardiol 2006;22:953-68.
24. Kostin S, Pool L, Elsässer A, et al. Myocytes die by multiple mechanisms in failing human hearts. Circ Res 2003;92:715-24.
25. Narumi T, Watanabe T, Kadowaki S, et al. Sarcopenia evaluated by fat-free mass index is an important prognostic factor in patients with chronic heart failure. Eur J Intern Med 2015;26:118-22.
26. Anker SD, Ponikowski P, Varney S, et al. Wasting as independent risk factor for mortality in chronic heart failure. Lancet 1997;349:1050-3.
27. Konishi M, Kagiyama N, Kamiya K, et al. Impact of sarcopenia on prognosis in patients with heart failure with reduced and preserved ejection fraction. Eur J Prev Cardiol 2021;28:1022-9.
28. Landi F, Marzetti E, Martone AM, Bernabei R, Onder G. Exercise as a remedy for sarcopenia. Curr Opin Clin Nutr Metab Care 2014;17:25-31.
29. Loncar G, Springer J, Anker M, Doehner W, Lainscak M. Cardiac cachexia: hic et nunc: “hic et nunc” - here and now. Int J Cardiol 2015;201:e1-12.
30. Anker SD, Negassa A, Coats AJS, et al. Prognostic importance of weight loss in chronic heart failure and the effect of treatment with angiotensin-converting-enzyme inhibitors: an observational study. Lancet 2003;361:1077-83.
31. Del Buono MG, Arena R, Borlaug BA, et al. Exercise intolerance in patients with heart failure: JACC state-of-the-art review. J Am Coll Cardiol 2019;73:2209-25.
32. von Haehling S, Ebner N, Dos Santos MR, Springer J, Anker SD. Muscle wasting and cachexia in heart failure: mechanisms and therapies. Nat Rev Cardiol 2017;14:323-41.
33. Piepoli MF, Davos C, Francis DP, Coats AJ. ExTraMATCH Collaborative. Exercise training meta-analysis of trials in patients with chronic heart failure (ExTraMATCH). BMJ 2004;328:189.
34. Anker SD, Chua TP, Ponikowski P, et al. Hormonal changes and catabolic/anabolic imbalance in chronic heart failure and their importance for cardiac cachexia. Circulation 1997;96:526-34.
35. Yamamoto E, Kato T, Yaku H, et al. Appetite loss at discharge from acute decompensated heart failure: observation from KCHF registry. PLoS One 2022;17:e0267327.
36. The 2019 American Geriatrics Society Beers Criteria® Update Expert Panel. American geriatrics society 2019 updated AGS beers criteria® for Potentially inappropriate medication use in older adults. J Am Geriatr Soc 2019;67:674-94.
37. Rocha de Avila Pelozin B, Felipe Rodrigues L, Menezes De Oliveira E, Fernandes T. Cardiac and cancer-associated cachexia: role of exercise training, non-coding RNAs, and future perspectives. In: D’Onofrio G, Cseri J, editors. Frailty and sarcopenia - recent evidence and new perspectives. IntechOpen; 2022.
38. Bruggeman AR, Kamal AH, LeBlanc TW, Ma JD, Baracos VE, Roeland EJ. Cancer cachexia: beyond weight loss. J Oncol Pract 2016;12:1163-71.
39. Musolino V, Palus S, Tschirner A, et al. Megestrol acetate improves cardiac function in a model of cancer cachexia-induced cardiomyopathy by autophagic modulation. J Cachexia Sarcopenia Muscle 2016;7:555-66.
40. Fernández-Pombo A, Rodríguez-Carnero G, Castro AI, et al. Relevance of nutritional assessment and treatment to counteract cardiac cachexia and sarcopenia in chronic heart failure. Clin Nutr 2021;40:5141-55.
41. Habaybeh D, de Moraes MB, Slee A, Avgerinou C. Nutritional interventions for heart failure patients who are malnourished or at risk of malnutrition or cachexia: a systematic review and meta-analysis. Heart Fail Rev 2021;26:1103-18.
42. Maimónides Biomedical Research Institute of Córdoba. Mediterranean diet and nutritional support in heart failure sarcopenia. 2023. Available from: https://clinicaltrials.gov/study/NCT05848960 [Last accessed on 21 Dec 2023].
43. Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res 2008;102:488-96.
44. Sanders PM, Russell ST, Tisdale MJ. Angiotensin II directly induces muscle protein catabolism through the ubiquitin-proteasome proteolytic pathway and may play a role in cancer cachexia. Br J Cancer 2005;93:425-34.
45. Lee JY, Kim DA, Choi E, Lee YS, Park SJ, Kim BJ. Aldosterone inhibits in vitro myogenesis by increasing intracellular oxidative stress via mineralocorticoid receptor. Endocrinol Metab 2021;36:865-74.
46. Burton LA, McMurdo MET, Struthers AD. Mineralocorticoid antagonism: a novel way to treat sarcopenia and physical impairment in older people? Clin Endocrinol 2011;75:725-9.
47. Angelidis G, Valotassiou V, Georgoulias P. Current and potential roles of ghrelin in clinical practice. J Endocrinol Invest 2010;33:823-38.
48. Akamizu T, Kangawa K. Therapeutic applications of ghrelin to cachexia utilizing its appetite-stimulating effect. Peptides 2011;32:2295-300.
49. Xin X, Ren AJ, Zheng X, et al. Disturbance of circulating ghrelin and obestatin in chronic heart failure patients especially in those with cachexia. Peptides 2009;30:2281-5.
50. Lenk K, Palus S, Schur R, et al. Effect of ghrelin and its analogues, BIM-28131 and BIM-28125, on the expression of myostatin in a rat heart failure model. J Cachexia Sarcopenia Muscle 2013;4:63-9.
51. Stitt TN, Drujan D, Clarke BA, et al. The IGF-1/PI3K/akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 2004;14:395-403.
52. Nagaya N, Moriya J, Yasumura Y, et al. Effects of ghrelin administration on left ventricular function, exercise capacity, and muscle wasting in patients with chronic heart failure. Circulation 2004;110:3674-9.
53. Zhang G, Yin X, Qi Y, et al. Ghrelin and cardiovascular diseases. Curr Cardiol Rev 2010;6:62-70.
55. Caminiti G, Volterrani M, Iellamo F, et al. Effect of long-acting testosterone treatment on functional exercise capacity, skeletal muscle performance, insulin resistance, and baroreflex sensitivity in elderly patients with chronic heart failure a double-blind, placebo-controlled, randomized study. J Am Coll Cardiol 2009;54:919-27.
56. Pugh PJ, Jones RD, West JN, Jones TH, Channer KS. Testosterone treatment for men with chronic heart failure. Heart 2004;90:446-7.
57. Mirdamadi A, Garakyaraghi M, Pourmoghaddas A, Bahmani A, Mahmoudi H, Gharipour M. Beneficial effects of testosterone therapy on functional capacity, cardiovascular parameters, and quality of life in patients with congestive heart failure. Biomed Res Int 2014;2014:392432.
58. Dos Santos MR, Sayegh ALC, Bacurau AVN, et al. Effect of exercise training and testosterone replacement on skeletal muscle wasting in patients with heart failure with testosterone deficiency. Mayo Clin Proc 2016;91:575-86.
59. Stout M, Tew GA, Doll H, et al. Testosterone therapy during exercise rehabilitation in male patients with chronic heart failure who have low testosterone status: a double-blind randomized controlled feasibility study. Am Heart J 2012;164:893-901.
60. Navarro-Peñalver M, Perez-Martinez MT, Gómez-Bueno M, et al. Testosterone replacement therapy in deficient patients with chronic heart failure: a randomized double-blind controlled pilot study. J Cardiovasc Pharmacol Ther 2018;23:543-50.
61. Malkin CJ, Pugh PJ, West JN, van Beek EJR, Jones TH, Channer KS. Testosterone therapy in men with moderate severity heart failure: a double-blind randomized placebo controlled trial. Eur Heart J 2006;27:57-64.
62. Lincoff AM, Bhasin S, Flevaris P, et al. Cardiovascular safety of testosterone-replacement therapy. N Engl J Med 2023;389:107-17.
63. Sculthorpe N, Solomon AM, Sinanan ACM, Bouloux PMG, Grace F, Lewis MP. Androgens affect myogenesis in vitro and increase local IGF-1 expression. Med Sci Sports Exerc 2012;44:610-5.
64. Ferrando AA, Tipton KD, Doyle D, Phillips SM, Cortiella J, Wolfe RR. Testosterone injection stimulates net protein synthesis but not tissue amino acid transport. Am J Physiol 1998;275:E864-71.
65. Powers ML, Florini JR. A direct effect of testosterone on muscle cells in tissue culture. Endocrinology 1975;97:1043-7.
66. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell 2017;168:960-76.
67. Adhikari R, Jha K, Dardari Z, et al. National trends in use of sodium-glucose cotransporter-2 inhibitors and glucagon-like peptide-1 receptor agonists by cardiologists and other specialties, 2015 to 2020. J Am Heart Assoc 2022;11:e023811.
68. Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: executive summary: a report of the american college of cardiology/American heart association joint committee on clinical practice guidelines. J Am Coll Cardiol 2022;79:1757-80.
69. Massimino E, Izzo A, Riccardi G, Della Pepa G. The impact of glucose-lowering drugs on sarcopenia in type 2 diabetes: current evidence and underlying mechanisms. Cells 2021;10:1958.
70. Dyck JRB, Sossalla S, Hamdani N, et al. Cardiac mechanisms of the beneficial effects of SGLT2 inhibitors in heart failure: evidence for potential off-target effects. J Mol Cell Cardiol 2022;167:17-31.
71. Takada S, Sabe H, Kinugawa S. Treatments for skeletal muscle abnormalities in heart failure: sodium-glucose transporter 2 and ketone bodies. Am J Physiol Heart Circ Physiol 2022;322:H117-28.
72. Rutledge C, Enriquez A, Redding K, et al. Liraglutide protects against diastolic dysfunction and improves ventricular protein translation. Cardiovasc Drugs Ther 2023.
73. Moore PW, Malone K, VanValkenburg D, et al. GLP-1 agonists for weight loss: pharmacology and clinical implications. Adv Ther 2023;40:723-42.
74. Wang JY, Wang QW, Yang XY, et al. GLP-1 receptor agonists for the treatment of obesity: role as a promising approach. Front Endocrinol 2023;14:1085799.
75. Gurjar AA, Kushwaha S, Chattopadhyay S, et al. Long acting GLP-1 analog liraglutide ameliorates skeletal muscle atrophy in rodents. Metabolism 2020;103:154044.
76. Hong Y, Lee JH, Jeong KW, Choi CS, Jun HS. Amelioration of muscle wasting by glucagon-like peptide-1 receptor agonist in muscle atrophy. J Cachexia Sarcopenia Muscle 2019;10:903-18.
77. Monzel AS, Enríquez JA, Picard M. Multifaceted mitochondria: moving mitochondrial science beyond function and dysfunction. Nat Metab 2023;5:546-62.
78. Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest 2018;128:3716-26.
79. Lai L, Leone TC, Keller MP, et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail 2014;7:1022-31.
80. Sagar S, Gustafsson AB. Cardiovascular aging: the mitochondrial influence. J Cardiovasc Aging 2023;3:33.
81. Mishra P, Varuzhanyan G, Pham AH, Chan DC. Mitochondrial dynamics is a distinguishing feature of skeletal muscle fiber types and regulates organellar compartmentalization. Cell Metab 2015;22:1033-44.
82. Adhihetty PJ, O'Leary MFN, Chabi B, Wicks KL, Hood DA. Effect of denervation on mitochondrially mediated apoptosis in skeletal muscle. J Appl Physiol 2007;102:1143-51.
83. Romanello V, Guadagnin E, Gomes L, et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J 2010;29:1774-85.
84. Coen PM, Musci RV, Hinkley JM, Miller BF. Mitochondria as a target for mitigating sarcopenia. Front Physiol 2019;9:01883.
85. Mau T, Blackwell TL, Cawthon PM, et al. Muscle mitochondrial bioenergetic capacities is associated with multimorbidity burden in older adults: the study of muscle, mobility and aging (SOMMA). 2023. Available from: https://www.x-mol.net/paper/detail/1721994890604990464 [Last accessed on 21 Dec 2023].
86. Min K, Smuder AJ, Kwon OS, Kavazis AN, Szeto HH, Powers SK. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J Appl Physiol 2011;111:1459-66.
87. McDonagh B, Scullion SM, Vasilaki A, Pollock N, McArdle A, Jackson MJ. Ageing-induced changes in the redox status of peripheral motor nerves imply an effect on redox signalling rather than oxidative damage. Free Radic Biol Med 2016;94:27-35.
88. Lv J, Li Y, Shi S, et al. Skeletal muscle mitochondrial remodeling in heart failure: an update on mechanisms and therapeutic opportunities. Biomed Pharmacother 2022;155:113833.
91. van der Ent M, Jeneson JA, Remme WJ, Berger R, Ciampricotti R, Visser F. A non-invasive selective assessment of type I fibre mitochondrial function using 31PNMR spectroscopy: evidence for impaired oxidative phosphorylation rate in skeletal muscle in patients with chronic heart failure. Eur Heart J 1998;19:124-31.
92. Doenst T, Pytel G, Schrepper A, et al. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc Res 2010;86:461-70.
93. Knapp F, Niemann B, Li L, et al. Differential effects of right and left heart failure on skeletal muscle in rats. J Cachexia Sarcopenia Muscle 2020;11:1830-49.
94. Schrepper A, Schwarzer M, Schöpe M, Amorim PA, Doenst T. Biphasic response of skeletal muscle mitochondria to chronic cardiac pressure overload - role of respiratory chain complex activity. J Mol Cell Cardiol 2012;52:125-35.
95. Acin-Perez R, Benincá C, Shabane B, Shirihai OS, Stiles L. Utilization of human samples for assessment of mitochondrial bioenergetics: gold standards, limitations, and future perspectives. Life 2021;11:949.
96. Guzmán Mentesana G, Báez AL, Lo Presti MS, et al. Functional and structural alterations of cardiac and skeletal muscle mitochondria in heart failure patients. Arch Med Res 2014;45:237-46.
97. Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res 2014;114:524-37.
98. Powers SK, Ji LL, Kavazis AN, Jackson MJ. Reactive oxygen species: impact on skeletal muscle. Compr Physiol 2011;1:941-69.
99. Jang JY, Blum A, Liu J, Finkel T. The role of mitochondria in aging. J Clin Invest 2018;128:3662-70.
100. Pinti M, Cevenini E, Nasi M, et al. Circulating mitochondrial DNA increases with age and is a familiar trait: implications for “inflamm-aging”. Eur J Immunol 2014;44:1552-62.
101. Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell 2014;54:289-96.
102. West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol 2017;17:363-75.
103. Shadel GS, Clayton DA. Mitochondrial DNA maintenance in vertebrates. Annu Rev Biochem 1997;66:409-35.
104. Ide T, Tsutsui H, Hayashidani S, et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res 2001;88:529-35.
105. Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res 2010;106:1541-8.
106. Hiona A, Leeuwenburgh C. The role of mitochondrial DNA mutations in aging and sarcopenia: implications for the mitochondrial vicious cycle theory of aging. Exp Gerontol 2008;43:24-33.
107. Mettauer B, Zoll J, Garnier A, Ventura-Clapier R. Heart failure: a model of cardiac and skeletal muscle energetic failure. Pflugers Arch 2006;452:653-66.
108. Bețiu AM, Noveanu L, Hâncu IM, et al. Mitochondrial effects of common cardiovascular medications: the good, the bad and the mixed. Int J Mol Sci 2022;23:13653.
109. Zoll J, Monassier L, Garnier A, et al. ACE inhibition prevents myocardial infarction-induced skeletal muscle mitochondrial dysfunction. J Appl Physiol 2006;101:385-91.
110. Bacurau AV, Cunha TF, Souza RW, Voltarelli VA, Gabriel-Costa D, Brum PC. Aerobic exercise and pharmacological therapies for skeletal myopathy in heart failure: similarities and differences. Oxid Med Cell Longev 2016;2016:4374671.
111. Casuso RA, Huertas JR. The emerging role of skeletal muscle mitochondrial dynamics in exercise and ageing. Ageing Res Rev 2020;58:101025.
112. Niemann B, Chen Y, Issa H, Silber RE, Rohrbach S. Caloric restriction delays cardiac ageing in rats: role of mitochondria. Cardiovasc Res 2010;88:267-76.
113. No MH, Heo JW, Yoo SZ, et al. Effects of aging and exercise training on mitochondrial function and apoptosis in the rat heart. Pflugers Arch 2020;472:179-93.
114. Peoples JN, Saraf A, Ghazal N, Pham TT, Kwong JQ. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med 2019;51:1-13.
115. Ussher JR, Jaswal JS, Lopaschuk GD. Pyridine nucleotide regulation of cardiac intermediary metabolism. Circ Res 2012;111:628-41.
116. Walker MA, Tian R. Raising NAD in heart failure: time to translate? Circulation 2018;137:2274-7.
117. Prolla TA, Denu JM. NAD+ deficiency in age-related mitochondrial dysfunction. Cell Metab 2014;19:178-80.
118. Henning RH, Brundel BJJM. Proteostasis in cardiac health and disease. Nat Rev Cardiol 2017;14:637-53.
119. Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol 2019;20:421-35.
120. Hetz C, Chevet E, Oakes SA. Proteostasis control by the unfolded protein response. Nat Cell Biol 2015;17:829-38.
121. Rutledge CA, Lagranha C, Chiba T, et al. Metformin preconditioning protects against myocardial stunning and preserves protein translation in a mouse model of cardiac arrest. J Mol Cell Cardiol Plus 2023;4:100034.
122. Meijering RAM, Zhang D, Hoogstra-Berends F, Henning RH, Brundel BJJM. Loss of proteostatic control as a substrate for atrial fibrillation: a novel target for upstream therapy by heat shock proteins. Front Physiol 2012;3:36. Available from: https://www.frontiersin.org/articles/10.3389/fphys.2012.00036 [Last accessed on 21 Dec 2023].
123. Christians ES, Benjamin IJ. Proteostasis and REDOX state in the heart. Am J Physiol Heart Circ Physiol 2012;302:H24-37.
124. Mainali N, Ayyadevara S, Ganne A, Shmookler Reis RJ, Mehta JL. Protein homeostasis in the aged and diseased heart. J Cardiovasc Aging 2023;3:16.
125. Paez HG, Pitzer CR, Alway SE. Age-related dysfunction in proteostasis and cellular quality control in the development of sarcopenia. Cells 2023;12:249.
126. Wu M, Falasca M, Blough ER. Akt/protein kinase B in skeletal muscle physiology and pathology. J Cell Physiol 2011;226:29-36.
127. Steinberg GR, Hardie DG. New insights into activation and function of the AMPK. Nat Rev Mol Cell Biol 2023;24:255-72.
128. Calvert JW, Gundewar S, Jha S, et al. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes 2008;57:696-705.
129. Bujak AL, Crane JD, Lally JS, et al. AMPK activation of muscle autophagy prevents fasting-induced hypoglycemia and myopathy during aging. Cell Metab 2015;21:883-90.
130. O’Neill HM. AMPK and exercise: glucose uptake and insulin sensitivity. Diabetes Metab J 2013;37:1-21.
131. Cao Y, Bojjireddy N, Kim M, et al. Activation of γ2-AMPK suppresses ribosome biogenesis and protects against myocardial ischemia/reperfusion injury. Circ Res 2017;121:1182-91.
132. Sorrenti V, Benedetti F, Buriani A, et al. Immunomodulatory and antiaging mechanisms of resveratrol, rapamycin, and metformin: focus on mTOR and AMPK signaling networks. Pharmaceuticals 2022;15:912.
133. Ziaaldini MM, Marzetti E, Picca A, Murlasits Z. Biochemical pathways of sarcopenia and their modulation by physical exercise: a narrative review. Front Med 2017;4:167.
134. Pearson G, Robinson F, Beers Gibson T, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 2001;22:153-83.
135. Basualto-Alarcón C, Jorquera G, Altamirano F, Jaimovich E, Estrada M. Testosterone signals through mTOR and androgen receptor to induce muscle hypertrophy. Med Sci Sports Exerc 2013;45:1712-20.
136. Cabello-Verrugio C, Cordova G, Salas JD. Angiotensin II: role in skeletal muscle atrophy. Curr Protein Pept Sci 2012;13:560-9.
137. Bossola M, Pacelli F, Costelli P, Tortorelli A, Rosa F, Doglietto GB. Proteasome activities in the rectus abdominis muscle of young and older individuals. Biogerontology 2008;9:261-8.
138. Whitman SA, Wacker MJ, Richmond SR, Godard MP. Contributions of the ubiquitin-proteasome pathway and apoptosis to human skeletal muscle wasting with age. Pflugers Arch 2005;450:437-46.
139. Centner T, Yano J, Kimura E, et al. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol 2001;306:717-26.
140. Chen SN, Czernuszewicz G, Tan Y, et al. Human molecular genetic and functional studies identify TRIM63, encoding muscle RING finger protein 1, as a novel gene for human hypertrophic cardiomyopathy. Circ Res 2012;111:907-19.
141. Bodine SC, Latres E, Baumhueter S, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001;294:1704-8.
142. Altun M, Besche HC, Overkleeft HS, et al. Muscle wasting in aged, sarcopenic rats is associated with enhanced activity of the ubiquitin proteasome pathway. J Biol Chem 2010;285:39597-608.
143. Maejima Y, Usui S, Zhai P, et al. Muscle-specific RING finger 1 negatively regulates pathological cardiac hypertrophy through downregulation of calcineurin A. Circ Heart Fail 2014;7:479-90.
144. Patterson C, Willis MS, Portbury A. Rise above: muscle ring-finger-1 (MURF1) regulation of cardiomyocyte size and energy metabolism. Trans Am Clin Climatol Assoc 2011;122:70-81.
145. Willis MS, Schisler JC, Li L, et al. Cardiac muscle ring finger-1 increases susceptibility to heart failure in vivo. Circ Res 2009;105:80-8.
146. Mangner N, Weikert B, Bowen TS, et al. Skeletal muscle alterations in chronic heart failure: differential effects on quadriceps and diaphragm. J Cachexia Sarcopenia Muscle 2015;6:381-90.
147. Gielen S, Sandri M, Kozarez I, et al. Exercise training attenuates MuRF-1 expression in the skeletal muscle of patients with chronic heart failure independent of age: the randomized Leipzig Exercise Intervention in Chronic Heart Failure and Aging catabolism study. Circulation 2012;125:2716-27.
148. Souza RWA, Piedade WP, Soares LC, et al. Aerobic exercise training prevents heart failure-induced skeletal muscle atrophy by anti-catabolic, but not anabolic actions. PLoS One 2014;9:e110020.
149. Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA 2001;98:14440-5.
150. Dehoux MJM, van Beneden RP, Fernández-Celemín L, Lause PL, Thissen JPM. Induction of MafBx and Murf ubiquitin ligase mRNAs in rat skeletal muscle after LPS injection. FEBS Lett 2003;544:214-7.
151. Adams V, Linke A, Wisloff U, et al. Myocardial expression of Murf-1 and MAFbx after induction of chronic heart failure: effect on myocardial contractility. Cardiovasc Res 2007;73:120-9.
152. Razeghi P, Baskin KK, Sharma S, et al. Atrophy, hypertrophy, and hypoxemia induce transcriptional regulators of the ubiquitin proteasome system in the rat heart. Biochem Biophys Res Commun 2006;342:361-4.
153. Carvalho RF, Castan EP, Coelho CA, et al. Heart failure increases atrogin-1 and MuRF1 gene expression in skeletal muscle with fiber type-specific atrophy. J Mol Histol 2010;41:81-7.
154. Santoyo-Suarez MG, Mares-Montemayor JD, Padilla-Rivas GR, et al. The involvement of krüppel-like factors in cardiovascular diseases. Life 2023;13:420.
155. Xu M, Chen X, Chen D, Yu B, Huang Z. FoxO1: a novel insight into its molecular mechanisms in the regulation of skeletal muscle differentiation and fiber type specification. Oncotarget 2017;8:10662-74.
156. Hirata Y, Nomura K, Kato D, et al. A piezo1/KLF15/IL-6 axis mediates immobilization-induced muscle atrophy. J Clin Invest 2022;132:1-13.
157. Gan Z, Fu T, Kelly DP, Vega RB. Skeletal muscle mitochondrial remodeling in exercise and diseases. Cell Res 2018;28:969-80.
158. Prosdocimo DA, Sabeh MK, Jain MK. Kruppel-like factors in muscle health and disease. Trends Cardiovasc Med 2015;25:278-87.
159. Pollak NM, Hoffman M, Goldberg IJ, Drosatos K. Krüppel-like factors: crippling and un-crippling metabolic pathways. JACC Basic Transl Sci 2018;3:132-56.
160. Sweet DR, Fan L, Hsieh PN, Jain MK. Krüppel-like factors in vascular inflammation: mechanistic insights and therapeutic potential. Front Cardiovasc Med 2018;5:6.
161. He C, Sumpter R Jr, Levine B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 2012;8:1548-51.
162. Grumati P, Coletto L, Schiavinato A, et al. Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles. Autophagy 2011;7:1415-23.
163. Sanchez AMJ, Csibi A, Raibon A, et al. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J Cell Biochem 2012;113:695-710.
164. Grumati P, Coletto L, Sabatelli P, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 2010;16:1313-20.
165. Fujita N, Fujino H, Sakamoto H, Takegaki J, Deie M. Time course of ubiquitin-proteasome and macroautophagy-lysosome pathways in skeletal muscle in rats with heart failure. Biomed Res 2015;36:383-92.
166. Jannig PR, Moreira JBN, Bechara LRG, et al. Autophagy signaling in skeletal muscle of infarcted rats. PLoS One 2014;9:e85820.
167. Adams V, Jiang H, Yu J, et al. Apoptosis in skeletal myocytes of patients with chronic heart failure is associated with exercise intolerance. J Am Coll Cardiol 1999;33:959-65.
169. Xie G, Jin H, Mikhail H, et al. Autophagy in sarcopenia: possible mechanisms and novel therapies. Biomed Pharmacother 2023;165:115147.
170. Conraads VM, Bosmans JM, Vrints CJ. Chronic heart failure: an example of a systemic chronic inflammatory disease resulting in cachexia. Int J Cardiol 2002;85:33-49.
171. Krysztofiak H, Wleklik M, Migaj J, et al. Cardiac cachexia: a well-known but challenging complication of heart failure. Clin Interv Aging 2020;15:2041-51.
172. Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med 1990;323:236-41.
173. Clark DJ, Cleman MW, Pfau SE, et al. Serum complement activation in congestive heart failure. Am Heart J 2001;141:684-90.
174. Rauchhaus M, Doehner W, Francis DP, et al. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation 2000;102:3060-7.
175. Tsutamoto T, Hisanaga T, Wada A, et al. Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J Am Coll Cardiol 1998;31:391-8.
177. Bozkurt B, Kribbs SB, Clubb FJ Jr, et al. Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation 1998;97:1382-91.
178. Li X, Moody MR, Engel D, et al. Cardiac-specific overexpression of tumor necrosis factor-alpha causes oxidative stress and contractile dysfunction in mouse diaphragm. Circulation 2000;102:1690-6.
179. Schaap LA, Pluijm SMF, Deeg DJH, et al. Higher inflammatory marker levels in older persons: associations with 5-year change in muscle mass and muscle strength. J Gerontol A Biol Sci Med Sci 2009;64:1183-9.
180. Dirks AJ, Leeuwenburgh C. Tumor necrosis factor alpha signaling in skeletal muscle: effects of age and caloric restriction. J Nutr Biochem 2006;17:501-8.
181. Costelli P, Carbó N, Tessitore L, et al. Tumor necrosis factor-alpha mediates changes in tissue protein turnover in a rat cancer cachexia model. J Clin Invest 1993;92:2783-9.
182. Nakamura K, Fushimi K, Kouchi H, et al. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circulation 1998;98:794-9.
183. Suematsu N, Tsutsui H, Wen J, et al. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation 2003;107:1418-23.
184. Morley JE, Thomas DR, Wilson MMG. Cachexia: pathophysiology and clinical relevance. Am J Clin Nutr 2006;83:735-43.
185. Testa M, Yeh M, Lee P, et al. Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J Am Coll Cardiol 1996;28:964-71.
186. Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol 1994;12:141-79.
187. Sartori R, Romanello V, Sandri M. Mechanisms of muscle atrophy and hypertrophy: implications in health and disease. Nat Commun 2021;12:330.
188. Todorov P, Cariuk P, McDevitt T, Coles B, Fearon K, Tisdale M. Characterization of a cancer cachectic factor. Nature 1996;379:739-42.
189. Tsujinaka T, Ebisui C, Fujita J, et al. Muscle undergoes atrophy in association with increase of lysosomal cathepsin activity in interleukin-6 transgenic mouse. Biochem Biophys Res Commun 1995;207:168-74.
190. Anker SD, Ponikowski PP, Clark AL, et al. Cytokines and neurohormones relating to body composition alterations in the wasting syndrome of chronic heart failure. Eur Heart J 1999;20:683-93.
191. Martins T, Vitorino R, Moreira-Gonçalves D, Amado F, Duarte JA, Ferreira R. Recent insights on the molecular mechanisms and therapeutic approaches for cardiac cachexia. Clin Biochem 2014;47:8-15.
192. Ronnebaum SM, Patterson C. The FoxO family in cardiac function and dysfunction. Annu Rev Physiol 2010;72:81-94.
193. Moylan JS, Smith JD, Chambers MA, McLoughlin TJ, Reid MB. TNF induction of atrogin-1/MAFbx mRNA depends on Foxo4 expression but not AKT-Foxo1/3 signaling. Am J Physiol Cell Physiol 2008;295:C986-93.
194. Rodríguez-Nuevo A, Zorzano A. The sensing of mitochondrial DAMPs by non-immune cells. Cell Stress 2019;3:195-207.
195. Zheng Y, Xu L, Dong N, Li F. NLRP3 inflammasome: the rising star in cardiovascular diseases. Front Cardiovasc Med 2022;9:927061.
196. Anker SD, Egerer KR, Volk HD, Kox WJ, Poole-Wilson PA, Coats AJS. Elevated soluble CD14 receptors and altered cytokines in chronic heart failure. Am J Cardiol 1997;79:1426-30.
197. Liu CF, Tang WHW. Gut microbiota in sarcopenia and heart failure. J Cardiovasc Aging 2022;2:35.
198. Ferrari R, Bachetti T, Agnoletti L, Comini L, Curello S. Endothelial function and dysfunction in heart failure. Eur Heart J 1998;19 Suppl G:G41-7.
199. Dos Santos MR, Saitoh M, Ebner N, et al. Sarcopenia and endothelial function in patients with chronic heart failure: results from the studies investigating comorbidities aggravating heart failure (SICA-HF). J Am Med Dir Assoc 2017;18:240-5.
200. Wilson JR, Martin JL, Schwartz D, Ferraro N. Exercise intolerance in patients with chronic heart failure: role of impaired nutritive flow to skeletal muscle. Circulation 1984;69:1079-87.
201. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol 2007;7:803-15.
202. Mann DL, McMurray JJV, Packer M, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 2004;109:1594-602.
203. Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT. Anti-TNF Therapy Against Congestive Heart Failure Investigators. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003;107:3133-40.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Rutledge CA. Molecular mechanisms underlying sarcopenia in heart failure. J Cardiovasc Aging 2024;4:7. http://dx.doi.org/10.20517/jca.2023.40
AMA Style
Rutledge CA. Molecular mechanisms underlying sarcopenia in heart failure. The Journal of Cardiovascular Aging. 2024; 4(1): 7. http://dx.doi.org/10.20517/jca.2023.40
Chicago/Turabian Style
Rutledge, Cody A.. 2024. "Molecular mechanisms underlying sarcopenia in heart failure" The Journal of Cardiovascular Aging. 4, no.1: 7. http://dx.doi.org/10.20517/jca.2023.40
ACS Style
Rutledge, CA. Molecular mechanisms underlying sarcopenia in heart failure. J. Cardiovasc. Aging. 2024, 4, 7. http://dx.doi.org/10.20517/jca.2023.40
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 8 clicks
Cite This Article 8 clicks

 Like This Article 5
likes
Like This Article 5
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.