
Lipid overload - a culprit for hypertrophic cardiomyopathy?

 ,
, Hypertrophic cardiomyopathy (HCM) is characterized by unexplained left ventricular hypertrophy in the absence of loading conditions such as hypertension or valvular diseases. It is the most common cause of inherited cardiac diseases, with a prevalence of 1 in 500 worldwide[1]. It is also the most common cause of sudden death in the young[2]. While etiology is heterogenous, at least half of the HCMs with a molecular diagnosis were due to pathogenic or likely pathogenic variants in sarcomere protein encoding genes, predominantly MYH7 (encoding beta-myosin heavy chain) and MYBPC3 (encoding cardiac myosin binding protein C)[2]. While the pathophysiology of HCM has been heavily debated, the most recent studies converge on the hypercontractility hypothesis[3]. Contractile protein mutations that cause either increased contractility or impaired relaxation can result in HCM. The main mechanisms that have been proposed are associated with increased calcium sensitivity, increased myosin head ATPase activity, or reduced myosin super-relaxed state among others. This hypothesis has been well supported by studies utilizing the disease-modeling murine models carrying human pathogenic variants or the patient-derived human induced pluripotent stem cell differentiated cardiomyocytes models[4]. The concentric hypertrophy associated with HCM has been attributed to increased calcium induced calcineurin signaling and activation of mitogen-activated protein kinase kinase 1-extracellular signal-regulated kinase 1/2 (MEK1-ERK1/2) signaling pathways[5]. In addition to MYH7 and MYBPC3 mutations, TNNI3 (encoding cardiac troponin I, cTnI) is another gene frequently associated with HCM. cTnI is the inhibitory subunit of the troponin complex and serves as a calcium-sensitive molecular switch of the sarcomere contraction in the myocardium. Notably, TNNI variants have been reported in 2%-7% of the HCM cases[6]. Interestingly, pathogenic variants of TNNI3 have been associated with HCM, dilated cardiomyopathy, and restrictive cardiomyopathy, presumably due to their differential effect on contractility; however, detailed genotype-phenotype association has not been well established [7].
TNNI3 p.R186Q mutation was previously reported in multiple families with HCM and a large Chinese family with co-segregated HCM and atrial fibrillation (AF)[8,9]. In this issue of the Journal of Cardiovascular Aging, Guo et al. characterized the cardiac phenotype in the first Tnni3R186Q/R186Q knockin mouse model and investigated the molecular mechanisms underlying the development of HCM[10]. Unlike the HCM patients who usually carry heterozygous pathogenic mutation, the heterozygous Tnni3R186Q/+ mice exhibit relatively normal cardiac function and cardiac morphology. The homozygous Tnni3R186Q/R186Q mice exhibit hallmarks of HCM at the age of 10 months including thickening of left ventricular wall, enlarged cardiomyocytes, increased fibrosis, and elevated levels of hypertrophic markers, while the total protein levels of cTnI were unchanged. The overall life span was shorter in Tnni3R186Q/R186Q mice than in their wild-type littermates, although the cause of death was not detailed in this study. Utilizing mass spectrometry, they then discovered the differentially expressed proteins in the hearts of Tnni3R186Q/R186Q mice compared with the control hearts. Interestingly, enrichment pathway analysis revealed that many altered proteins were involved in fatty acid metabolism. Consistently, they showed that lipid droplets and the levels of total triglycerides and total cholesterol were increased in the hearts of Tnni3R186Q/R186Q mice. Because the plasma levels of lipid content remained unchanged, it suggests that the lipid overload in the hearts of Tnni3R186Q/R186Q mice is of cardiac origin. Furthermore, fatty acid synthase (FASN) - a key metabolic enzyme involved in the fatty acid metabolism pathway was found to be elevated in the Tnni3R186Q/R186Q hearts. They then focused on FASN as an intermediator between aberrant fatty acid metabolism and cardiac hypertrophy [Figure 1]. Strikingly, the dietary intake of the FASN-selective inhibitor C75 for an extended period (i.e., 2 months) improved the cardiac lipid profile, reduced lipid droplets, and attenuated the hypertrophic myopathy in Tnni3R186Q/R186Q mice.
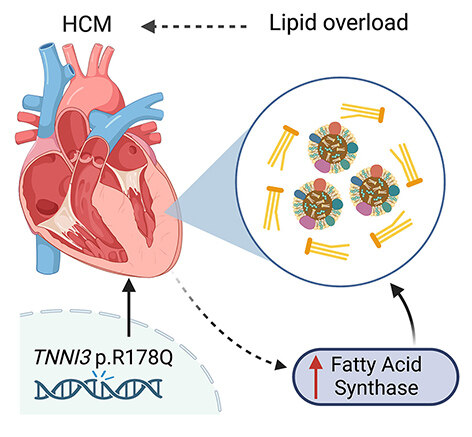
Figure 1. Lipid overload enhances HCM. HCM: Hypertrophic cardiomyopathy.
Defective lipid metabolism has not been scrutinized in HCM. However, increased contractility and the resulting higher energy demand must be supported by efficient metabolism. Inefficient energy utilization has been shown to cause energetic stress and adverse remodeling[11]. Two recent multi-omics studies have shown that energy metabolism is impaired in the heart of HCM patients [12,13]. In particular, increased levels of free fatty acids and reduced acylcarnitines, with broadly downregulated gene expression and protein reduction across fatty acids transport, activation, and β-oxidation, suggest a severely depressed fatty acid metabolism associated with HCM, similar to what have been reported in the advanced heart failure. Inborn errors of fatty acid metabolism and carnitine deficiencies are reported to provoke secondary HCM. The study by Guo et al. provides another surprising link between the altered fatty acid metabolism and HCM development[10]. The observed lipid accumulation in cTnI R186Q knockin hearts is most likely a result of enhanced fatty acid synthesis, rather than the impaired or slowed breakdown of fatty acids, as the proteins involved in the fatty acid β-oxidation pathway were overall unchanged. Interestingly, human studies on HCM hearts with both sarcomeric and non-sarcomeric variants did not reveal accumulation of triacylglyceride, i.e., lipid droplets[12,13]. Further previous animal models, including those with other cTnI variant transgenic models, which recapitulated phenotypic HCM, did not reveal a fatty acid accumulation phenotype[14]. Whether the lipid accumulation is unique to the current model or variant or is more broadly applicable requires further evaluation.
The impact of cTnI R186Q mutation on contraction and relaxation of sarcomeres was not directly evaluated in this study. It would be interesting to determine whether the current model represents an example of the hypercontractility hypothesis or an example of a storage disease, such as Fabry disease-associated HCM, which phenocopies sarcomeric HCM. Although the precise molecular mechanism needs to be further elucidated, the study by Guo et al. certainly provides new evidence supporting the link between the sarcomeric variant and lipid accumulation. Future investigations should also address whether restoring fatty acid metabolic homeostasis by reducing synthesis or enhancing fatty acid catabolism would be efficacious for HCM. Moreover, considering the recent success of Mavacamten, a cardiac myosin inhibitor for the obstructive HCM[15], it may be worth evaluating whether the patients with the cTnI R186Q mutation might be responsive to the Mavacamten treatment. These potential research questions not only can further provide insights into the molecular mechanisms underlying the cTnI R186Q associated HCM, but also could shed light on the therapeutic indications.
DECLARATIONS
Authors’ contributionsConceived and wrote the paper: Zhang L, Li N
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis study was supported by grants from National Institutes of Health (R01HL143067, R01HL150589, R21HL165270 to Zhang L, and R01HL136389 and R01HL163277, R01HL147108 to Li N), Li N received American Heart Association Established Investigator Award (936111).
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and regulatory approvalNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
2. Ho CY, Day SM, Ashley EA, et al. Genotype and ifetime burden of disease in hypertrophic cardiomyopathy: insights from the sarcomeric human cardiomyopathy registry (SHaRe). Circulation 2018;138:1387-98.
3. Yotti R, Seidman CE, Seidman JG. Advances in the genetic basis and pathogenesis of sarcomere cardiomyopathies. Annu Rev Genomics Hum Genet 2019;20:129-53.
4. Davis J, Davis LC, Correll RN, et al. A Tension-Based Model Distinguishes Hypertrophic versus Dilated Cardiomyopathy. Cell 2016;165:1147-59.
5. Kehat I, Davis J, Tiburcy M, et al. Extracellular signal-regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ Res 2011;108:176-83.
6. Pua CJ, Tham N, Chin CWL, et al. Genetic studies of hypertrophic cardiomyopathy in singaporeans identify variants in TNNI3 and TNNT2 That are common in Chinese patients. Circ Genom Precis Med 2020;13:424-34.
7. Mogensen J, Hey T, Lambrecht S. A Systematic review of phenotypic features associated with cardiac troponin I mutations in hereditary cardiomyopathies. Can J Cardiol 2015;31:1377-85.
8. Mogensen J, Murphy RT, Kubo T, et al. Frequency and clinical expression of cardiac troponin I mutations in 748 consecutive families with hypertrophic cardiomyopathy. J Am Coll Cardiol 2004;44:2315-25.
9. Hu J, Xiang Li J, Hong K, Xin Hu J, Brugada P, Shu Cheng X. Hypertrophic cardiomyopathy and planned in vitro fertilization. Genetic testing and clinical evaluation. Herz 2012;37:447-52.
10. Guo L, Su Y, Chen C, et al. The TNNI3 p.R186Q mutation is responsible for hypertrophic cardiomyopathy via promoting FASN-stimulated abnormal fatty acid metabolism. J Cardiovasc Aging 2023;3:6.
11. van der Velden J, Tocchetti CG, Varricchi G, et al. Metabolic changes in hypertrophic cardiomyopathies: scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc Res 2018;114:1273-80.
12. Previs MJ, O'Leary TS, Morley MP, et al. Defects in the Proteome and Metabolome in Human Hypertrophic Cardiomyopathy. Circ Heart Fail 2022;15:e009521.
13. Ranjbarvaziri S, Kooiker KB, Ellenberger M, et al. Altered cardiac energetics and mitochondrial dysfunction in hypertrophic cardiomyopathy. Circulation 2021;144:1714-31.
14. Wang Y, Pinto JR, Solis RS, et al. Generation and functional characterization of knock-in mice harboring the cardiac troponin I-R21C mutation associated with hypertrophic cardiomyopathy. J Biol Chem 2012;287:2156-67.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Zhang L, Li N. Lipid overload - a culprit for hypertrophic cardiomyopathy?. J Cardiovasc Aging 2023;3:8. http://dx.doi.org/10.20517/jca.2022.43
AMA Style
Zhang L, Li N. Lipid overload - a culprit for hypertrophic cardiomyopathy?. The Journal of Cardiovascular Aging. 2023; 3(1): 8. http://dx.doi.org/10.20517/jca.2022.43
Chicago/Turabian Style
Zhang, Lilei, Na Li. 2023. "Lipid overload - a culprit for hypertrophic cardiomyopathy?" The Journal of Cardiovascular Aging. 3, no.1: 8. http://dx.doi.org/10.20517/jca.2022.43
ACS Style
Zhang, L.; Li N. Lipid overload - a culprit for hypertrophic cardiomyopathy?. J. Cardiovasc. Aging. 2023, 3, 8. http://dx.doi.org/10.20517/jca.2022.43
About This Article
Copyright

Data & Comments
Data


 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 5
likes
Like This Article 5
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.