

The Journal of Cardiovascular Aging








Topic: Stem Cell & Genomics From Precision Medicine to Clinical Trial in Dish
NaN
Shelby A. Hall , Lisa A. Lesniewski
, Lisa A. Lesniewski
, Lisa A. Lesniewski
Views: Downloads:
Views: Downloads:
Pasquale Mone, ... Gaetano Santulli
Views: Downloads:
Meredith Whitehead, ... Catherine M. Shanahan
Views: Downloads:
Shah R. Ali, ... Hesham A. Sadek
, ... Hesham A. Sadek
Views: Downloads:
Leila Rouhi, ... Ali J. Marian
, ... Ali J. Marian
Views: Downloads:
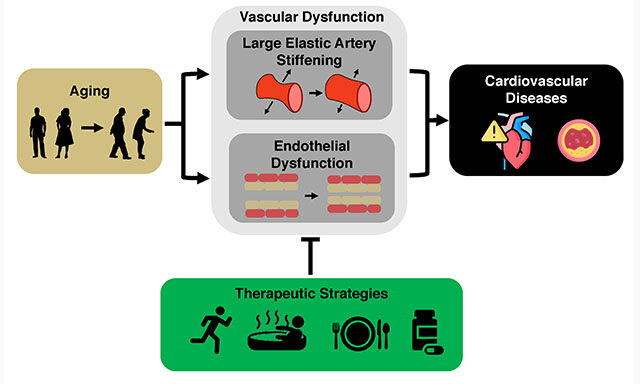
Zachary S. Clayton, ... Matthew J. Rossman
, ... Matthew J. Rossman
Views: Downloads:
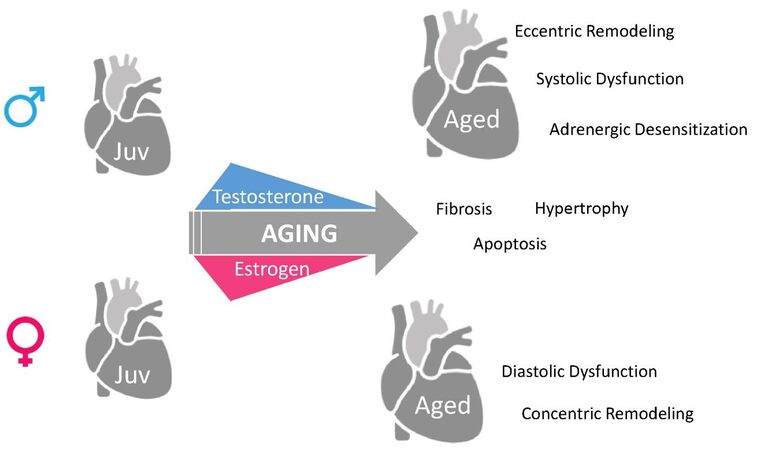
Aykhan Yusifov, ... Danielle R. Bruns
Views: Downloads:






Data
520
Authors
86
Reviewers
2021
Published Since
132
Citations
176,692
Article Views
74,542
Article Downloads
For Reviewers
For Readers
Add your e-mail address to receive forthcoming Issues of this journal:
Themed Collections
Topic: Stem Cell & Genomics From Precision Medicine to Clinical Trial in Dish
NaN
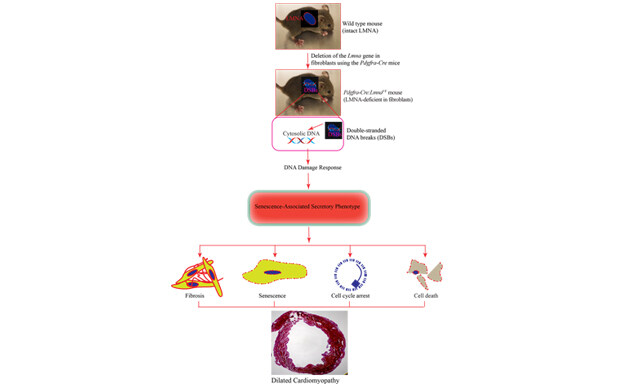
Leila Rouhi, ... Ali J. Marian
, ... Ali J. MarianOpen Access | Original Research Article | 9 Jun 2022
Views: | Downloads:
Data
520
Authors
86
Reviewers
2021
Published Since
132
Citations
176,692
Article Views
74,542
Article Downloads



