
Protein homeostasis in the aged and diseased heart

 , ...
, ... Abstract
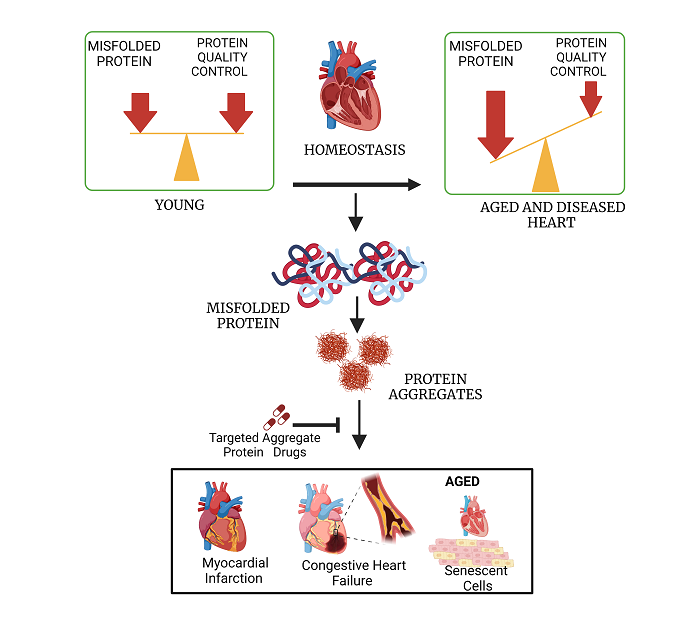
Protein homeostasis, the balance between protein synthesis and degradation, requires the clearance of misfolded and aggregated proteins and is therefore considered to be an essential aspect of establishing a physiologically effective proteome. Aging alters this balance, termed “proteostasis”, resulting in the progressive accumulation of misfolded and aggregated proteins. Defective proteostasis leads to the functional deterioration of diverse regulatory processes during aging and is implicated in the etiology of multiple pathological conditions underlying a variety of neurodegenerative diseases and in age-dependent cardiovascular disease. Detergent-insoluble protein aggregates have been reported by us in both aged and hypertensive hearts. The protein constituents were found to overlap with protein aggregates seen in neurodegenerative diseases such as Alzheimer’s disease. Therefore, targeting these protein components of aggregates may be a promising therapeutic strategy for cardiovascular pathologies associated with aging, ischemia, and/or hypertension.
Keywords
INTRODUCTION
Cardiovascular diseases (CVD) have been and remain the leading cause of death worldwide, and account for 23.5% of U.S. mortality (CDC 2017). Hypertension, high blood cholesterol, diabetes, obesity, smoking, physical inactivity, gender, age, and genetics are the leading contributors to CVDs. Disruption of protein homeostasis, accumulation of misfolded proteins, and protein aggregation have also been linked to several types of CVDs[1]. A deeper understanding of protein homeostasis and protein aggregation, and their impairment in diseased hearts, may enable the development of new strategies and therapeutic targets for CVD prevention and intervention.
Protein homeostasis is fundamental for proper cell function and survival. A healthy proteome contains both proteins that are properly folded into a well-structured native state, as well as many intrinsically disordered proteins which have regions lacking a stable structure, or which achieve stability only upon binding of one or more ligands, or in association with other proteins in dimeric or multimeric complexes. Whereas most proteins fold spontaneously, many proteins require molecular chaperones to assist in folding[2,3]. There are several monitoring systems in the cell that detect any proteins that have been misfolded, partially folded, or are intrinsically disordered or unstructured. Misfolded proteins have exposed hydrophobic regions that can adhere to similar regions in nearby proteins, leading to nonfunctional binding and aggregation. Accumulating such aberrant complexes was previously shown to be highly toxic to cells[4].
The accumulation of misfolded proteins in a cell or subcellular compartment elicits the Unfolded Protein Response (UPR), which recruits chaperones required for proper protein folding[5-8]. Accumulation of misfolded proteins in the endoplasmic reticulum (ER) triggers the ER-associated degradation (ERAD) pathway via glucose-regulated protein 78 (GRP78), inositol-requiring protein 1 (IRE1), activating transcription factor 6 (ATF-6), and PKR-like ER kinase (PERK). The UPR pathway helps to limit misfolding by halting or reducing protein synthesis at the endoplasmic reticulum[9-12]. The Ubiquitin-Proteasome System (UPS) chiefly handles misfolded proteins, whereas autophagy provides an alternative degradation route for degraded organelles and larger aggregates[13-16].
THE UBIQUITIN-PROTEASOME SYSTEM
UPS is a highly regulated process that degrades proteins, most of which are structurally aberrant (and thus potentially dysfunctional), due to misfolding, oxidation, excessive post-translational misfolding, and/or entanglement of intrinsically disordered proteins or regions. UPS machinery has been identified in nucleus and cytosol, and degrades proteins from all cell compartments including membranes, nucleus, cytoplasm, and the ER lumen. UPS is an ATP-dependent system comprising two complementary processes: ubiquitination and proteasomal degradation[17,18]. Ubiquitination occurs in three steps. First, a ubiquitin-activating enzyme (E1) binds ATP and transfers AMP-adenylate to a C-terminal carboxylate; it then captures that ubiquitin monomer through a Cys-thioester linkage and transfers it to carrier protein E2 via a thioester exchange. Ubiquitin ligases (E3) then catalyze substrate-specific ubiquitination of target proteins. There are over 600 substrate-specific E3 ligases[19], which recognize misfolded target proteins (or their associated chaperones) and catalyze the transfer of the ubiquitin moiety to proteins destined to be degraded after assembly of a polyubiquitin chain. Since ubiquitin modification is a dynamic process, not all polyubiquitinated proteins are destined for degradation; they also serve as signaling molecules that trigger DNA repair or activate kinases and stabilize certain proteins in cancer cells[20-22]. Polyubiquitinated proteins destined to be degraded are transferred to proteasomes, where they are deubiquitinated and cleaved into fragments of < 30 residues[23,24].
UPS in the mammalian heart is distinguished by several features. E3 ligases such as atrogin-1, the muscle ring finger (MuRF) family, and C-terminal HSP70-interacting protein (CHIP) assist in the conjugation of ubiquitin to substrates peculiar to or especially abundant in the heart[25]. All proteasomes consist of two sub-complexes: a 20S proteasome, also known as the catalytic core particle (CP), and one or two 19S proteasome activators, also known as regulatory particles (RPs). The CP comprises subpopulations of α and β catalytic subunits, which exist in constitutive forms (β1, β2, and β5), as well as immune forms (β1i, β2i, and β5i) that make up the immunoproteasome[26,27]. The 20S core of cardiac proteasomes contains a mixture of constitutive and immune proteasome subunits. The 19S regulatory particle may also be replaced by an 11S regulatory particle in mouse heart, complexed with the 20S catalytic core particle. The resulting 20S-11S complex appears to improve the catabolism of many cardiac substrates[17,28,29].
UPS IN THE DISEASED HEART
The relationship between abnormal UPS activity and pathogenesis was first recognized in diverse neurodegenerative diseases, including Alzheimer’s, Parkinson’s, and Huntington’s diseases. Subsequently, several studies have determined the role of UPS and UPS-related therapeutic targets in disease states of other organs, including the heart[30,31]. MDM2 (an E3 ligase) has been shown to target the p53 protein, which is both a tumor suppressor and an inducer of apoptosis, for ubiquitination and proteasomal processing[32,33]. Elevated levels of p53 and MDM2 have been observed in heart failure patients, suggesting that increased apoptosis may be directly related to UPS dysregulation in diseased hearts[34]. Several studies have reported that proteasome subunits can undergo post-translational modifications (PTM) or oxidative damage that inhibit their catabolic function in ischemia-reperfusion (IR) injury models, thus perhaps contributing to UPS dysfunction leading to pathologic hypertrophy during and after CVD[35-37]. Total ubiquitinated protein load has been found to increase in several cardiovascular pathologies[38-41]. Impairment in coupling of ubiquitination and proteasomal degradation has also been shown to be a contributing factor to models of myocardial ischemic-reperfusion injury in both yeast and mice[42,43].
AUTOPHAGY
Lysosomes are the key degradative organelles in autophagy, an intracellular “recycling” pathway that serves to eliminate large, defective cellular components including cell organelles (e.g., mitochondria) as well as pathogens and cytosolic aggregates. Such structures are too large to be degraded by proteasomes but can be cytotoxic if not eliminated. Recycling of damaged and/or harmful cellular components by degradation allows the recovery of ATP, amino acids, and nucleotides for cell metabolism. There are three main types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Previous studies have also discovered crosstalk between UPS and autophagy[44]. Under normal conditions or moderate stress, autophagy clears old and damaged cell organelles, and dysfunctional protein complexes, as a cytoprotective alternative to apoptosis. The balance between apoptosis and autophagy, essential for proper functioning of the heart, is maintained by mTOR and Endoplasmic Reticulum (ER) stress pathways but disrupted by rapamycin treatment[45]. Cardiomyocytes, specialized muscle cells that maintain the contraction-relaxation cycle of the heart, are rich in mitochondria - resulting in a dependence on mitochondrial autophagy (termed “mitophagy”) for achieving homeostasis.
AUTOPHAGY IN THE DISEASED HEART
Mortality of cardiac cells can arise via apoptosis, ischemic cell death, or autophagic cell death[46-49]. Mice with Atg5- or Atg7-deficient hearts are more susceptible to cardiac dysfunction induced by ER and mitochondrial stress than mice with wild-type hearts[50,51]. Cardiomyocytes in hearts from animal models of ischemia-reperfusion (I/R) injury have elevated abundance of autophagosomes, suggesting an impairment in their clearance after I/R injury[52]. Mice with myocardial infarction (MI) were found to have reduced autophagic flux and mitochondrial respiration relative to sham-operated mice. This decrease in flux results in an accumulation of damaged organelles, polyubiquitinated proteins, and spheroidal (more rounded) mitochondria[53,54]. Autophagy is an essential protective mechanism that fails during long-term heart dysfunction[49,55-57]. Oxidative damage to proteins required for autophagic functions (e.g., Atg4, Atg5, and Atg7) can lead to the failure of cardioprotection in CVD[58-61].
CROSSTALK AND COOPERATION BETWEEN UPS AND AUTOPHAGY
Inhibition of UPS proteasomes disrupts the ERAD pathway and increases ER stress markers (e.g., IRE1, ATF6, PERK). IRE1 activation results in recruitment of TRAF2 required for phosphorylation of JNK, and expression of autophagy-pathway genes. Upon cleavage, ATF6 translocates to the nucleus and triggers expression of DAPK1 and phosphorylation of beclin-1 required for autophagosome biogenesis. Also, activated PERK through eIF2alpha activates ATF4 and CHOP, in turn inducing expression of diverse autophagy (ATG) genes[62-66]. Inhibition of autophagy leads to compromised UPS-associated substrate protein clearance[67]. Shuttling proteins, such as SQSTM-1/p62, responsible for the proper conveyance of ubiquitinated substrates to the proteasome, also accumulate in aggregates due to the inhibition of autophagy[67,68].
UPS and autophagy also work together in the clearance of unnatural, misfolded, and unrequired proteins[26,69]. This has been well documented in Huntington’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (ALS), characterized by aggregation of their respective hallmark proteins such as Htt. α-synuclein and TDP-43. All these aggregates require both proteasomal and lysosomal degradation pathways[70,71]. Also, molecular chaperones such as CHIP and BAG have multiple functionalities. CHIP acts as a co-chaperone for HSP70 and HSP90 for proteasomal degradation, but can also mediate autophagic degradation of Lys63-specific misfolded proteins[72,73]. Among proteins in the BAG family, BAG1 mediates proteasomal degradation, while BAG3 cooperates with Hsp70, CHIP and SQSTM-1/p62 for autophagic degradation[74,75].
MOLECULAR MECHANISMS LEADING TO CVD
Myocardial ischemia and heart failure (HF) are the most common conditions that lead to CVD mortality. Each of these conditions initiates hypoxia, oxidative stress, ER and mitochondrial stress, and cardiac remodeling. Several studies have shown that a cascade of cellular-stress events can alter the machinery of UPS and/or autophagy, resulting in their disrupted clearance functions and the intracellular accumulation of misfolded proteins[38,69,75-79].
HYPOXIA (ISCHEMIA)
Organ-specific oxygen consumption depends on the type and condition of each tissue. The heart and brain create the highest demand for oxygen, and cardiac consumption rises sharply during vigorous exercise. Hypoxia refers to an oxygen level (partial pressure) that is below the physiological requirements of an organism or a tissue. Genes that are elicited in hypoxic environments are termed hypoxia-inducible and are upregulated by hypoxia-inducible factor-1α (HIF-1α) and HIF-2α acting as transcription factors or co-factors. In normoxic conditions, HIF-1α is hydroxylated through an oxygen-dependent process and is degraded through a UPS pathway. HIF-1α protein is stabilized in hypoxic conditions and translocates into the nucleus, where it binds to hypoxia-response elements (HREs) to activate the transcription of hypoxia-inducible genes. Genes directly targeted by HIF-1α are chiefly those responsible for glycolysis, angiogenesis, and erythropoiesis, but there are numerous secondary targets[80-82]. Hypoxia negatively impacts protein synthesis through such mechanisms as inhibition of mTOR signaling, and activation of PERK in the ER - both of which are responsible for regulation of translation initiation[83-86]. Hypoxia also leads to activation of UPR machinery. Hypoxia-induced phosphorylation of eIF2α activates ATF4, a transcription factor that regulates autophagy and expression of CHOP (which induces apoptosis) and GADD34 (involved in the integrated stress response pathway)[87,88]. Hypoxia also induces protein misfolding, especially in the ER, which is itself a trigger of inflammation pathways[88]. Disrupted UPR, an oxygen-dependent process, contributes to the accumulation of misfolded proteins in aggregates. Other branches of the ER-stress pathway, mediated by ATF6 and IRE1, are also activated by hypoxia[89,90]. Additional pathways impacted by hypoxia include apoptosis, cell-cycle regulation, DNA repair, and metabolism, as shown by microarray analysis comparing normoxic vs. hypoxic cells[91].
Decreased oxygen delivery, leading to tissue hypoxia, occurs in several cardiovascular diseases including atherosclerosis, hypertension, and heart failure. In atherosclerosis, monocytes recruited by inflammatory signals to the arterial wall differentiate into macrophages, whose nitric oxide (NO) production is enhanced by HIF-1α but opposed by HIF-2α[92,93]. Since NO is a primary mediator of inflammation, hypoxia is pro-inflammatory in atherosclerosis. HIF-1α has been shown to regulate hypertension in mice; HIF-1α+/- mice show a significant decrease in hypertension induced by chronic-intermittent hypoxia compared to wild-type mice[94,95]. In humans, chronic hypoxia causes activation of the sympathetic nervous system and increased systemic arterial pressure[96]. Prolonged hypoxia leads to activation of autophagy, which protects cardiomyocytes from ER stress and apoptosis[97,98]. Angiotensin receptor-neprilysin inhibitor (ARNI), a compound recently developed to treat heart failure, reduces hypoxia-induced myocardial injury through inhibition of autophagy[99].
OXIDATIVE STRESS
Elevated levels of free radicals, chiefly reactive oxygen species (ROS), result in the disruption of several cell-signaling and growth-mediating pathways. At low physiological levels, ROS play a critical role in diverse cellular processes including phosphorylation and activation of several transcription factors[100,101]. Protein carbonylation increases during oxidative stress, as observed for the 19S regulatory S6 proteasome subunit, considered to be the oxidation-sensitive subunit in 26S proteasomes[102]. Post-translational modifications related to oxidative stress, such as carbonylation, were shown to accumulate in proteins in aged mice[103]. Carbonylation of side-chain amino acids is linked to protein aggregation by promoting unfolding[104,105].
ROS are generated as metabolic by-products of oxidative phosphorylation by mitochondria, which are especially abundant in cardiomyocytes; however, they also originate from nitric oxide synthase (NOS) and diverse other enzymes such as NADPH oxidase (NOX) and cytochromes p450. Mitochondria play a dominant role in determining ROS levels, as they amplify NOX-derived ROS and thus function as redox hubs in cardiac physiology and disease. High ROS levels affect the disposition of myocardial calcium, induce arrhythmia, and can contribute to cardiac remodeling[106]. In addition to free radicals generated in the myocardium (cardiomyocytes), leukocytes produce a large proportion of cardiac ROS. Although small amounts of ROS are also generated under normal physiological conditions, the antioxidant defense mechanisms are often adequate to remove excess ROS and maintain oxidative balance. However, during hypoxia, the oxidant load surpasses the heart’s ability to counter the ROS load resulting in oxidative stress. ROS are highly reactive toward proteins, lipids, and DNA; oxidative damage to DNA leads to somatic mutations, the vast majority of which are deleterious, whereas their impact on proteins and membranes can result in cell dysfunction or death. Alterations in protein and calcium homeostasis due to oxidative stress have been previously implicated in the heart, where they are most readily observed through structural modifications to cardiomyocytes[107,108]. NOX4, a member of NADPH oxidase family of proteins, is responsible for transfer of electrons from either NADPH or NADH to molecular oxygen to generate
ER STRESS
The endoplasmic reticulum (ER) is a vital organelle responsible for essential cellular functions such as protein synthesis, protein folding, calcium homeostasis, and the generation of autophagosomes. Under normal physiological conditions, GRP78 (a key ER chaperone) remains bound to effector molecules such as ATF6, IRE1, and PERK; during stress, however, these effectors dissociate from GRP78 and become activated. Activated ATF6 is cleaved in Golgi bodies, releasing its N-terminal fragment that acts as a transcription factor to induce genes that protect against ER stress and ROS. Activated IRE1 splices mRNA encoding X-box binding protein 1 (XBP1); the spliced XBP1 isoform translocates to the nucleus to initiate transcription of molecular chaperones. Activated PERK phosphorylates eukaryotic initiation factor 2 alpha (eIF2α) which in turn activates ATF4 and then C/EBP homologous protein (CHOP)[118]. As discussed above, there is interplay and crosstalk between various pathways involved in ER stress, hypoxia, and UPS. Senstrin2 is an important regulator of cell responses to various stresses[119,120]. In a recent study, ER stress was shown to induce cardiac autophagy and myocardial dysfunction in Sestrin2-knockout mice[121].
Mild ER stress is beneficial for the cell, as it clears any unfolded or damaged proteins via an ER-specific UPR termed UPRER; however, prolonged ER stress leads to cell death and is cardiotoxic since it leads to apoptosis of cardiomyocytes[122]. Because cardiomyocytes cannot regenerate, extended periods of ER stress can cause irreparable damage to the heart. Since ER plays a vital role in the metabolism of lipids and the maturation and folding of transmembrane proteins, suppression of ER stress is critical to survival. ER stress nevertheless occurs in atherosclerotic plaques in humans and in animal models of atherosclerosis, although ameliorated by flavonoids[123-125].
MITOCHONDRIAL STRESS
Mitochondria are another essential cell organelle responsible for generating ATP as the principal energy currency required for cell metabolism and enabling cardiomyocyte contraction and relaxation. Other critical functions of mitochondria include biosynthetic metabolic processes, chiefly through the Krebs cycle, the maintenance of sustainable ROS levels, monitoring protein integrity, and preventing the accumulation of damaged proteins[118]. Under physiological stress, mitochondria activate their own UPR, termed UPRMit. This initiates transcription of the mitochondrial protease ClpP, mitochondrial chaperones or heat-shock proteins (HSP60 and HSP10), and proteins involved in ROS detoxification. The transcription of these factors is believed to be initiated by activated ATF5. Another stress-induced pathway is the activation of PERK and c-Jun N-terminal kinase 2 (JNK2), both of which participate in the translation of ATF4, CHOP, and ATF5 - proteins that contribute to cardiomyocyte dysfunction and apoptotic cell death. Mitochondrial stress also activates an autophagy-variant clearance pathway to remove dysfunctional mitochondria, called mitophagy[126,127]. Glutathione is involved in cellular homeostasis, protects cells from oxidative stress, and inhibits apoptosis[128]. Treatment with exogenous glutathione can help to restore mitochondrial redox status and eventually decrease mitochondrial stress leading to improvement in cardiovascular function of aged mice[129].
Crosstalk between mitochondria and ER has been observed and is believed to be a vital process in maintaining cardiac function. Calcium sequestered in ER is channeled to mitochondria, where it is required for ATP production. Proteins implicated in mitochondrial calcium uptake include Mfn2, VAPB, PTPIP51, and STIM1. STIM1 is an ER-resident calcium sensor; its genetic knockout in mice disrupts both ER and mitochondrial functions, leading to cardiac fibrosis and myopathy. Similar interactions between ER and mitochondria are implicated by autophagy dysregulation via JNK, Bcl-2, and Atg6[130-132]. Both mitochondrial and ER stress responses are tightly connected to other mechanisms involved in protein homeostasis, including UPS, autophagy, hypoxia, and oxidative stress during cardiac dysfunction; consequently, proteins involved in these stress responses might be valuable targets for therapeutic interventions.
CARDIAC REMODELING
Cardiac tissue is susceptible to damage after ischemic injury, or cardiomyopathy, arising from hypoxia, oxidative stress, ER, or mitochondrial stress. Changes in size, mass, geometry, and function of the heart, as well as its overall morphology and function, are observed after cardiac injury - changes collectively known as cardiac remodeling. Following ischemic injury, there is extensive death of cardiomyocytes. The necrotic tissue after MI is replaced by extracellular matrix (ECM), resulting in fibrosis. Cardiac fibroblasts have high plasticity and are involved in the generation of ECM, scar formation, and inter-cellular signaling[133-135]. They increase their collagen synthesis, causing fibrosis not only of infarct tissue but also of non-infarct areas, thus participating in further cardiac remodeling[136,137]. Although initial remodeling of the heart is important to compensate for damage incurred, prolonged remodeling leads to the deterioration of cardiac function and may culminate in heart failure[138]. Prolonged remodeling of the heart is associated with ventricular arrhythmias, increased neurohormonal activation, and excessive vasoconstriction[139,140]. Several treatment strategies to reverse cardiac remodeling after heart failure have been studied in recent years. Renin-angiotensin system modulators, beta-blockers, neprilysin inhibitors and aldosterone antagonists are some of those drugs shown to promote angiogenesis after MI and to alter cardiac remodeling[141,142].
RELATIONSHIP BETWEEN MYOCARDIAL ISCHEMIA, AGING, AND HYPERTENSION
Hypertension, a common consequence of vascular remodeling, is a major health problem that often accompanies aging. With increasing age, the arteries and blood vessels become stiffer, which increases vascular resistance. Elevated blood pressure leads to CVD, kidney disease, vascular dementia, and/or eye problems. We recently reported that detergent-insoluble protein aggregates, isolated from aging and hypertensive mouse hearts, increased several-fold relative to young, normal hearts[143]. About 50% of all aggregate proteins increased in abundance with aging, while 55% increased with hypertension; a shared 30% of aggregate proteins increased with both aging and hypertension[143]. Remarkably, one-fifth of these “overlap” aggregate proteins were previously implicated in age-related neurodegenerative diseases (e.g., Alzheimer’s disease) and CVDs[143-147]. Many of these proteins are involved in protein homeostasis and specifically in UPS, including proteasome α subunits, and NEDD4, an E3 ligase that increases in protein aggregates with aging and hypertension[143,148]. Similarly, TOMM22 and TOMM40, proteins involved in mitochondrial protein import, are enriched in hypertensive mouse hearts[143,149]. NADH dehydrogenase implicated in cardiotoxicity through oxidative stress is seen to be enriched in both aged and hypertensive mice hearts. Other proteins such as Serine peptidase inhibitor 1 (Serpinh1) and Prohibitin are enriched with aging and hypertension[143,150].
Myocardial infarction (MI) following prolonged ischemia is associated with cardiomyocyte death (necrosis or apoptosis)[151]. Post-MI cardiomyocyte death is followed by cardiac remodeling (heart chamber dilation, ventricular wall thinning, and loss of cardiac function). Several studies have reported that following MI, cardiomyocytes undergo an unfolded protein response (UPR) that leads to increased apoptosis[152]. Prolonged ischemia results in mitochondrial swelling and the release of cytochrome C, contributing to contractile dysfunction[153]. In the ER, ischemia leads to impaired protein folding due to abnormal activation and dysregulation of ER chaperones, contributing to protein misfolding, impaired calcium homeostasis, and apoptosis[154]. After simulating MI in wild-type mice with left coronary artery (LCA) ligation, we showed for the first time that detergent-insoluble aggregates increase ~2.7-fold after MI relative to sham mice (unpublished data). Of the cardiac proteins enriched after experimental MI, ~9% coincided with proteins previously observed in aged and hypertensive heart aggregates [Figure 1]. Proteins previously implicated in protein homeostasis failure associated with neurodegenerative and/or CVDs, such as CAND1, SerpinH1, NEDD4, UCP1, Plectin-1, 14-3-3[155], ApoE, Cardiac Phospholamban, and HSP-90[156], were shared in common between MI, aging, and hypertension — suggesting that these conditions accrue protein aggregates due to defects in the same or similar pathways [Figure 1].
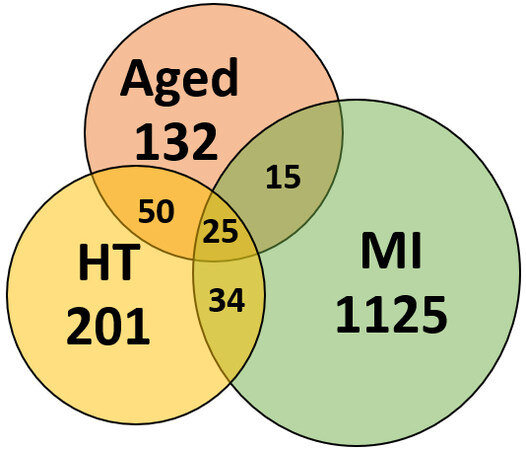
Figure 1. Proteins sequestered into aggregates overlap between aged, hypertensive (HT), and infarcted (MI) hearts.
The overlap among proteins enriched in cardiac aggregates due to aging, hypertension (HT), and experimental myocardial infarction (MI) implicates novel targets for therapeutic intervention.
THERAPEUTIC IMPLICATIONS OF IMPAIRED PROTEIN HOMEOSTASIS
Proteins involved in UPS, autophagy, mitochondrial and ER stress, and those directly affected by hypoxia and oxidative stress, are regulated by known pathways and suggest novel therapeutic interventions to mitigate CVDs. These proteins can either be upregulated, silenced, or otherwise manipulated to restore or maintain proteostatic equilibrium [Figure 2]. Drugs can be screened and evaluated for affinity to these proteins by methods employed previously[157-162], thus regulating their activity or interactions. Mitochondrial and ER proteins (e.g., PERK, IRE1, eiF2α, BiP, CHOP, GRP78, XBP1, ATF6, and ATF4) are known to be altered in diverse neurodegenerative diseases, and may be similarly aberrant in cardiac tissue, leading to protein aggregation and cardiomyopathy. In addition, autophagy regulators that include Atg4, Atg7, Atg3, and Atg10 have also been documented as reporters of CVD[163-165].
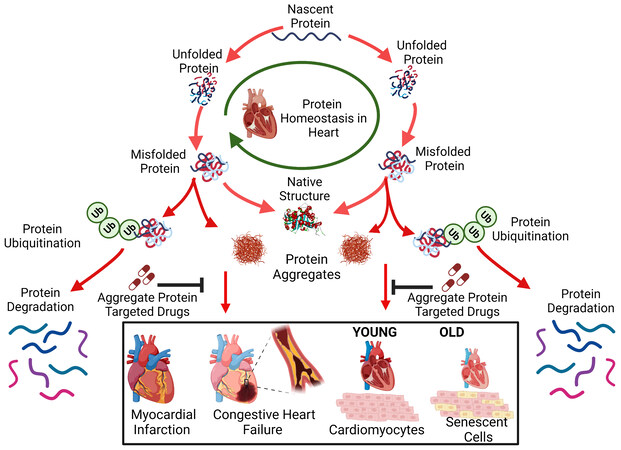
Figure 2. Drugs targeting cardiac-aggregate proteins can improve clearance of misfolded proteins and restore protein homeostasis compromised by cardiovascular disease (such as ischemia) and/or aging.
Proteins may misfold during translation or subsequently due to oxidation, other damage, or excessive post-translational modification. Misfolded proteins may be refolded with the help of chaperones, but if they remain in a disordered state, they are vulnerable to aggregation. Individual proteins or complexes can be cleared by the ubiquitin-proteasome system, but if not, then they can only be eliminated via autophagy.
Additional proteins specific to myocardial ischemia and related conditions, as well as those shared with hypertension and aging, may be implicated as contributors to abnormal proteostasis. Understanding protein homeostasis pathways and how aggregated protein components contribute to aggregate accumulation and consequent functional decline are novel areas of cardiovascular research. Unconventional methods developed to combine proteomics and interactome analyses have allowed us to identify proteins and protein-protein interfaces that are especially influential for aggregation. These provide novel targets for the discovery of drugs that oppose aggregation [Figure 2] as described recently[166].
Since dysfunctions of proteasomes and autophagy have been implicated in aging and age-related diseases, with consequences as diverse as those of Alzheimer’s disease and CVD, small molecules targeting influential aggregate proteins may also benefit those afflicted with a wide range of other age-associated conditions[167,168]. Targeting these aggregate-specific proteins with small molecules rescued aggregate-associated functional declines in AD models[155,157,160,166,169]. In view of the substantial overlaps between proteins sequestered into aggregates in aged, hypertensive and post-MI hearts, these aggregate-specific proteins and their interfaces (especially the shared protein sets) offer attractive drug targets to relieve protein aggregation and its associated physiological impairments. Therapies that improve cardiac function impaired by cardiac aging or by specific CVD events, especially myocardial ischemia, may target proteins defective in multiple pathologies, and thus serve as therapeutic approaches with the potential to ameliorate a wide range of age-related diseases.
DECLARATIONS
Authors’ contributionsWriting: Mainali N, Ayyadevara S
Additional contributions: Ganne A, Shmookler Reis RJ, Mehta JL
Availability of data and materialsNot applicable.
Financial support and sponsorshipWe are grateful for funding support as follows: NIH R01 (1R01AG062254) to Ayyadevara S. and Shmookler Reis R.J.; Merit grant (I01BX001655) and Senior Research Career Scientist Award to Shmookler Reis R.J. from the U.S. Dept. of Veteran Affairs; Program Project Grant 2P01AG012411-17A1 (W.S.T. Griffin, P.I.) from the National Institute on Aging (NIA/NIH); and Merit Review Award (I01 BX000282) and BRAVE funding award (14-052) to Mehta J.L. from the U.S. Department of Veterans Affairs.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Monte F, Agnetti G. Protein post-translational modifications and misfolding: new concepts in heart failure. Proteomics Clin Appl 2014;8:534-42.
4. Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med 2003;81:678-99.
6. Lebeaupin C, Vallée D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol 2018;69:927-47.
7. Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 2014;16:495-501.
8. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016;529:326-35.
9. Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol 2008;9:944-57.
10. Needham PG, Brodsky JL. How early studies on secreted and membrane protein quality control gave rise to the ER associated degradation (ERAD) pathway: the early history of ERAD. Biochim Biophys Acta 2013;1833:2447-57.
11. Bonifacino JS, Cosson P, Klausner RD. Colocalized transmembrane determinants for ER degradation and subunit assembly explain the intracellular fate of TCR chains. Cell 1990;63:503-13.
12. Ren J, Bi Y, Sowers JR, Hetz C, Zhang Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat Rev Cardiol 2021;18:499-521.
13. Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol 1998;143:1883-98.
14. García-Mata R, Bebök Z, Sorscher EJ, Sztul ES. Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J Cell Biol 1999;146:1239-54.
15. Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy 2008;4:141-50.
16. Reiss Y, Gur E, Ravid T. Releasing the lockdown: an emerging role for the ubiquitin-proteasome system in the breakdown of transient protein inclusions. Biomolecules 2020;10:1168.
17. Day SM. The ubiquitin proteasome system in human cardiomyopathies and heart failure. Am J Physiol Heart Circ Physiol 2013;304:H1283-93.
18. Li Y, Li S, Wu H. Ubiquitination-proteasome system (UPS) and autophagy two main protein degradation machineries in response to cell stress. Cells 2022;11:851.
19. George AJ, Hoffiz YC, Charles AJ, Zhu Y, Mabb AM. A comprehensive atlas of E3 ubiquitin ligase mutations in neurological disorders. Front Genet 2018;9:29.
20. Sun L, Chen ZJ. The novel functions of ubiquitination in signaling. Curr Opin Cell Biol 2004;16:119-26.
21. Brinkmann K, Schell M, Hoppe T, Kashkar H. Regulation of the DNA damage response by ubiquitin conjugation. Front Genet 2015;6:98.
22. Diefenbacher M, Orian A. Stabilization of nuclear oncoproteins by RNF4 and the ubiquitin system in cancer. Mol Cell Oncol 2017;4:e1260671.
23. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 2002;82:373-428.
24. Grice GL, Nathan JA. The recognition of ubiquitinated proteins by the proteasome. Cell Mol Life Sci 2016;73:3497-506.
25. Willis MS, Bevilacqua A, Pulinilkunnil T, Kienesberger P, Tannu M, Patterson C. The role of ubiquitin ligases in cardiac disease. J Mol Cell Cardiol 2014;71:43-53.
26. Wang C, Wang X. The interplay between autophagy and the ubiquitin-proteasome system in cardiac proteotoxicity. Biochim Biophys Acta 2015;1852:188-94.
27. Griffin TA, Nandi D, Cruz M, et al. Immunoproteasome assembly: cooperative incorporation of interferon gamma (IFN-gamma)-inducible subunits. J Exp Med 1998;187:97-104.
28. Li J, Powell SR, Wang X. Enhancement of proteasome function by PA28α overexpression protects against oxidative stress. FASEB J 2011;25:883-93.
29. Li J, Horak KM, Su H, Sanbe A, Robbins J, Wang X. Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J Clin Invest 2011;121:3689-700.
30. Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 2006;17:1807-19.
32. Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem 2000;275:8945-51.
33. Toth A, Nickson P, Qin LL, Erhardt P. Differential regulation of cardiomyocyte survival and hypertrophy by MDM2, an E3 ubiquitin ligase. J Biol Chem 2006;281:3679-89.
34. Birks EJ, Latif N, Enesa K, et al. Elevated p53 expression is associated with dysregulation of the ubiquitin-proteasome system in dilated cardiomyopathy. Cardiovasc Res 2008;79:472-80.
35. Scruggs SB, Zong NC, Wang D, Stefani E, Ping P. Post-translational modification of cardiac proteasomes: functional delineation enabled by proteomics. Am J Physiol Heart Circ Physiol 2012;303:H9-18.
37. Predmore JM, Wang P, Davis F, et al. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 2010;121:997-1004.
38. Wang X, Robbins J. Proteasomal and lysosomal protein degradation and heart disease. J Mol Cell Cardiol 2014;71:16-24.
39. Liu J, Chen Q, Huang W, et al. Impairment of the ubiquitin-proteasome system in desminopathy mouse hearts. FASEB J 2006;20:362-4.
40. Depre C, Wang Q, Yan L, et al. Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation 2006;114:1821-8.
41. Chen Q, Liu JB, Horak KM, et al. Intrasarcoplasmic amyloidosis impairs proteolytic function of proteasomes in cardiomyocytes by compromising substrate uptake. Circ Res 2005;97:1018-26.
42. Lu K, den Brave F, Jentsch S. Receptor oligomerization guides pathway choice between proteasomal and autophagic degradation. Nat Cell Biol 2017;19:732-9.
43. Hu C, Tian Y, Xu H, et al. Inadequate ubiquitination-proteasome coupling contributes to myocardial ischemia-reperfusion injury. J Clin Invest 2018;128:5294-306.
44. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol 2010;221:3-12.
45. Gao G, Chen W, Yan M, et al. Rapamycin regulates the balance between cardiomyocyte apoptosis and autophagy in chronic heart failure by inhibiting mTOR signaling. Int J Mol Med 2020;45:195-209.
46. Anversa P, Leri A, Beltrami CA, Guerra S, Kajstura J. Myocyte death and growth in the failing heart. Lab Invest 1998;78:767-86.
47. Narula J, Haider N, Virmani R, et al. Apoptosis in myocytes in end-stage heart failure. N Engl J Med 1996;335:1182-9.
48. Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 1995;146:3-15.
49. Ikeda S, Zablocki D, Sadoshima J. The role of autophagy in death of cardiomyocytes. J Mol Cell Cardiol 2022;165:1-8.
50. Collier JJ, Suomi F, Oláhová M, McWilliams TG, Taylor RW. Emerging roles of ATG7 in human health and disease. EMBO Mol Med 2021;13:e14824.
51. Nakai A, Yamaguchi O, Takeda T, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 2007;13:619-24.
52. Ma X, Liu H, Foyil SR, et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation 2012;125:3170-81.
53. Evans S, Ma X, Wang X, et al. Targeting the autophagy-lysosome pathway in a pathophysiologically relevant murine model of reversible heart failure. JACC Basic Transl Sci 2022;7:1214-28.
54. Wu P, Yuan X, Li F, et al. Myocardial upregulation of cathepsin d by ischemic heart disease promotes autophagic flux and protects against cardiac remodeling and heart failure. Circ Heart Fail 2017:10.
55. Shimomura H, Terasaki F, Hayashi T, Kitaura Y, Isomura T, Suma H. Autophagic degeneration as a possible mechanism of myocardial cell death in dilated cardiomyopathy. Jpn Circ J 2001;65:965-8.
56. Nishino I, Fu J, Tanji K, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000;406:906-10.
57. Nah J, Zablocki D, Sadoshima J. The role of autophagic cell death in cardiac disease. J Mol Cell Cardiol 2022;173:16-24.
58. Morales CR, Pedrozo Z, Lavandero S, Hill JA. Oxidative stress and autophagy in cardiovascular homeostasis. Antioxid Redox Signal 2014;20:507-18.
59. Luo C, Li Y, Wang H, et al. Mitochondrial accumulation under oxidative stress is due to defects in autophagy. J Cell Biochem 2013;114:212-9.
60. Terman A, Dalen H, Eaton JW, Neuzil J, Brunk UT. Aging of cardiac myocytes in culture: oxidative stress, lipofuscin accumulation, and mitochondrial turnover. Ann N Y Acad Sci 2004;1019:70-7.
61. Wang Y, Lu X, Wang X, et al. atg7-Based autophagy activation reverses doxorubicin-induced cardiotoxicity. Circ Res 2021;129:e166-82.
62. B'chir W, Maurin AC, Carraro V, et al. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 2013;41:7683-99.
63. Wang J, Kang R, Huang H, et al. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 2014;10:766-84.
64. Deegan S, Saveljeva S, Gorman AM, Samali A. Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cell Mol Life Sci 2013;70:2425-41.
65. Gade P, Ramachandran G, Maachani UB, et al. An IFN-γ-stimulated ATF6-C/EBP-β-signaling pathway critical for the expression of death associated protein kinase 1 and induction of autophagy. Proc Natl Acad Sci USA 2012;109:10316-21.
66. Zalckvar E, Berissi H, Eisenstein M, Kimchi A. Phosphorylation of beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 2009;5:720-2.
67. Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 2009;33:517-27.
68. Tian Z, Wang C, Hu C, Tian Y, Liu J, Wang X. AUTOPHAGIC-lysosomal inhibition compromises ubiquitin-proteasome system performance in a p62 dependent manner in cardiomyocytes. PLoS One 2014;9:e100715.
69. Zheng Q, Li J, Wang X. Interplay between the ubiquitin-proteasome system and autophagy in proteinopathies. Int J Physiol Pathophysiol Pharmacol 2009;1:127-42.
70. Cha-Molstad H, Sung KS, Hwang J, et al. Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat Cell Biol 2015;17:917-29.
71. Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med 2015;47:e147.
72. Blessing NA, Brockman AL, Chadee DN. The E3 ligase CHIP mediates ubiquitination and degradation of mixed-lineage kinase 3. Mol Cell Biol 2014;34:3132-43.
73. Zhang HT, Zeng LF, He QY, Tao WA, Zha ZG, Hu CD. The E3 ubiquitin ligase CHIP mediates ubiquitination and proteasomal degradation of PRMT5. Biochim Biophys Acta 2016;1863:335-46.
74. Cristofani R, Crippa V, Rusmini P, et al. Inhibition of retrograde transport modulates misfolded protein accumulation and clearance in motoneuron diseases. Autophagy 2017;13:1280-303.
75. Pan B, Li J, Parajuli N, et al. The Calcineurin-TFEB-p62 pathway mediates the activation of cardiac macroautophagy by proteasomal malfunction. Circ Res 2020;127:502-18.
76. Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J 2012;441:523-40.
77. Martins-Marques T, Ribeiro-Rodrigues T, Pereira P, Codogno P, Girao H. Autophagy and ubiquitination in cardiovascular diseases. DNA Cell Biol 2015;34:243-51.
78. Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J. Aging and autophagy in the heart. Circ Res 2016;118:1563-76.
79. Sciarretta S, Maejima Y, Zablocki D, Sadoshima J. The role of autophagy in the heart. Annu Rev Physiol 2018;80:1-26.
80. Lee SH, Golinska M, Griffiths JR. HIF-1-independent mechanisms regulating metabolic adaptation in hypoxic cancer cells. Cells 2021;10:2371.
81. Zimna A, Kurpisz M. Hypoxia-inducible factor-1 in physiological and pathophysiological angiogenesis: applications and therapies. Biomed Res Int 2015;2015:549412.
82. Kierans SJ, Taylor CT. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol 2021;599:23-37.
83. Koumenis C, Naczki C, Koritzinsky M, et al. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol 2002;22:7405-16.
84. Arsham AM, Howell JJ, Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem 2003;278:29655-60.
85. Brugarolas J, Lei K, Hurley RL, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 2004;18:2893-904.
86. Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell 2006;21:521-31.
87. Fels DR, Koumenis C. The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol Ther 2006;5:723-8.
88. Díaz-Bulnes P, Saiz ML, López-Larrea C, Rodríguez RM. Crosstalk between hypoxia and ER stress response: a key regulator of macrophage polarization. Front Immunol 2019;10:2951.
89. Jia W, Jian Z, Li J, et al. Upregulated ATF6 contributes to chronic intermittent hypoxia-afforded protection against myocardial ischemia/reperfusion injury. Int J Mol Med 2016;37:1199-208.
90. Cao X, He Y, Li X, Xu Y, Liu X. The IRE1α-XBP1 pathway function in hypoxia-induced pulmonary vascular remodeling, is upregulated by quercetin, inhibits apoptosis and partially reverses the effect of quercetin in PASMCs. Am J Transl Res 2019;11:641-54.
91. Blais JD, Filipenko V, Bi M, et al. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol 2004;24:7469-82.
92. Olson N, van der Vliet A. Interactions between nitric oxide and hypoxia-inducible factor signaling pathways in inflammatory disease. Nitric Oxide 2011;25:125-37.
93. Woollard KJ, Geissmann F. Monocytes in atherosclerosis: subsets and functions. Nat Rev Cardiol 2010;7:77-86.
94. Peng YJ, Yuan G, Ramakrishnan D, et al. Heterozygous HIF-1alpha deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol 2006;577:705-16.
95. Nanduri J, Peng YJ, Yuan G, Kumar GK, Prabhakar NR. Hypoxia-inducible factors and hypertension: lessons from sleep apnea syndrome. J Mol Med 2015;93:473-80.
96. Calbet JA. Chronic hypoxia increases blood pressure and noradrenaline spillover in healthy humans. J Physiol 2003;551:379-86.
97. Przyklenk K, Dong Y, Undyala VV, Whittaker P. Autophagy as a therapeutic target for ischaemia/reperfusion injury? Cardiovasc Res 2012;94:197-205.
98. Liu X, Deng Y, Xu Y, Jin W, Li H. MicroRNA-223 protects neonatal rat cardiomyocytes and H9c2 cells from hypoxia-induced apoptosis and excessive autophagy via the Akt/mTOR pathway by targeting PARP-1. J Mol Cell Cardiol 2018;118:133-46.
99. An J, Wang W, Guo Y, Wang C, Bao Q, Jia Y. Angiotensin receptor-neprilysin inhibitor attenuates ischemia-hypoxia-induced myocardial injury via inhibition of autophagy. Am J Transl Res 2022;14:8611-20.
100. Pizzino G, Irrera N, Cucinotta M, et al. Oxidative stress: harms and benefits for human health. Oxid Med Cell Longev 2017;2017:8416763.
101. Rajendran P, Nandakumar N, Rengarajan T, et al. Antioxidants and human diseases. Clin Chim Acta 2014;436:332-47.
102. Ishii T, Sakurai T, Usami H, Uchida K. Oxidative modification of proteasome: identification of an oxidation-sensitive subunit in 26 S proteasome. Biochemistry 2005;44:13893-901.
103. Tanase M, Urbanska AM, Zolla V, et al. Role of carbonyl modifications on aging-associated protein aggregation. Sci Rep 2016;6:19311.
104. Cannizzo ES, Clement CC, Morozova K, et al. Age-related oxidative stress compromises endosomal proteostasis. Cell Rep 2012;2:136-49.
105. Scharf B, Clement CC, Yodmuang S, et al. Age-related carbonylation of fibrocartilage structural proteins drives tissue degenerative modification. Chem Biol 2013;20:922-34.
106. Münzel T, Camici GG, Maack C, Bonetti NR, Fuster V, Kovacic JC. Impact of oxidative stress on the heart and vasculature: part 2 of a 3-part series. J Am Coll Cardiol 2017;70:212-29.
107. Breitkreuz M, Hamdani N. A change of heart: oxidative stress in governing muscle function? Biophys Rev 2015;7:321-41.
108. Lee A, Jeong D, Mitsuyama S, et al. The role of SUMO-1 in cardiac oxidative stress and hypertrophy. Antioxid Redox Signal 2014;21:1986-2001.
109. Serrander L, Cartier L, Bedard K, et al. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J 2007;406:105-14.
110. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245-313.
111. Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res 2010;106:1253-64.
112. Ago T, Liu T, Zhai P, et al. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell 2008;133:978-93.
113. Ikeda Y, Sciarretta S, Nagarajan N, et al. New insights into the role of mitochondrial dynamics and autophagy during oxidative stress and aging in the heart. Oxid Med Cell Longev 2014;2014:210934.
114. Li D, Yang Y, Wang S, et al. Role of acetylation in doxorubicin-induced cardiotoxicity. Redox Biol 2021;46:102089.
115. Wallace KB. Adriamycin-induced interference with cardiac mitochondrial calcium homeostasis. Cardiovasc Toxicol 2007;7:101-7.
116. Prathumsap N, Shinlapawittayatorn K, Chattipakorn SC, Chattipakorn N. Effects of doxorubicin on the heart: from molecular mechanisms to intervention strategies. Eur J Pharmacol 2020;866:172818.
117. Chen X, Zhang H, Feng M, Xu Z, Qian L. A novel peptide HSP-17 ameliorates oxidative stress injury and apoptosis in H9c2 cardiomyocytes by activating the PI3K/Akt pathway. Ann Transl Med 2022;10:1357.
118. Li J, Zhang D, Brundel BJJM, Wiersma M. Imbalance of ER and mitochondria interactions: prelude to cardiac ageing and disease? Cells 2019;8:1617.
119. Ren D, Quan N, Fedorova J, Zhang J, He Z, Li J. Sestrin2 modulates cardiac inflammatory response through maintaining redox homeostasis during ischemia and reperfusion. Redox Biol 2020;34:101556.
120. Kishimoto Y, Kondo K, Momiyama Y. The protective role of sestrin2 in atherosclerotic and cardiac diseases. Int J Mol Sci 2021;22:1200.
121. Zhang J, Yao L, Li S, Ferdous M, Zhao P. ER stress induces myocardial dysfunction and cardiac autophagy in Sestrin2 knockout mice. Am J Transl Res 2022;14:5800-11.
122. Okada K, Minamino T, Tsukamoto Y, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 2004;110:705-12.
123. Liu MQ, Chen Z, Chen LX. Endoplasmic reticulum stress: a novel mechanism and therapeutic target for cardiovascular diseases. Acta Pharmacol Sin 2016;37:425-43.
124. Giordano E, Davalos A, Nicod N, Visioli F. Hydroxytyrosol attenuates tunicamycin-induced endoplasmic reticulum stress in human hepatocarcinoma cells. Mol Nutr Food Res 2014;58:954-62.
125. Keylani K, Arbab Mojeni F, Khalaji A, et al. Endoplasmic reticulum as a target in cardiovascular diseases: is there a role for flavonoids? Front Pharmacol 2022;13:1027633.
126. Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci 2004;117:4055-66.
127. Moyzis AG, Sadoshima J, Gustafsson ÅB. Mending a broken heart: the role of mitophagy in cardioprotection. Am J Physiol Heart Circ Physiol 2015;308:H183-92.
128. Marí M, Morales A, Colell A, García-Ruiz C, Fernández-Checa JC. Mitochondrial glutathione, a key survival antioxidant. Antioxid Redox Signal 2009;11:2685-700.
129. Strutynska N, Goshovska Y, Mys L, et al. Glutathione restores the mitochondrial redox status and improves the function of the cardiovascular system in old rats. Front Physiol 2022;13:1093388.
130. Chen Y, Csordás G, Jowdy C, et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ Res 2012;111:863-75.
131. Gordaliza-Alaguero I, Cantó C, Zorzano A. Metabolic implications of organelle-mitochondria communication. EMBO Rep 2019;20:e47928.
132. Collins HE, He L, Zou L, et al. Stromal interaction molecule 1 is essential for normal cardiac homeostasis through modulation of ER and mitochondrial function. Am J Physiol Heart Circ Physiol 2014;306:H1231-9.
134. Tomasek JJ, McRae J, Owens GK, Haaksma CJ. Regulation of alpha-smooth muscle actin expression in granulation tissue myofibroblasts is dependent on the intronic CArG element and the transforming growth factor-beta1 control element. Am J Pathol 2005;166:1343-51.
135. Daseke MJ 2nd, Tenkorang MAA, Chalise U, Konfrst SR, Lindsey ML. Cardiac fibroblast activation during myocardial infarction wound healing: Fibroblast polarization after MI. Matrix Biol 2020;91-92:109-16.
136. van Krimpen C, Smits JF, Cleutjens JP, et al. DNA synthesis in the non-infarcted cardiac interstitium after left coronary artery ligation in the rat: effects of captopril. J Mol Cell Cardiol 1991;23:1245-53.
137. Volders PG, Willems IE, Cleutjens JP, Arends JW, Havenith MG, Daemen MJ. Interstitial collagen is increased in the non-infarcted human myocardium after myocardial infarction. J Mol Cell Cardiol 1993;25:1317-23.
138. Azevedo PS, Polegato BF, Minicucci MF, Paiva SA, Zornoff LA. Cardiac remodeling: concepts, clinical impact, pathophysiological mechanisms and pharmacologic treatment. Arq Bras Cardiol 2016;106:62-9.
139. Cohn JN, Levine TB, Olivari MT, et al. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med 1984;311:819-23.
140. Hartupee J, Mann DL. Neurohormonal activation in heart failure with reduced ejection fraction. Nat Rev Cardiol 2017;14:30-8.
141. Yang P, Li X, Wang L, et al. Effects of sacubitril/valsartan on cardiac reverse remodeling and cardiac resynchronization in patients with acute myocardial infarction. Front Cardiovasc Med 2022;9:1059420.
142. Zhang MX, Huang XY, Song Y, Xu WL, Li YL, Li C. Astragalus propinquus schischkin and Salvia miltiorrhiza bunge promote angiogenesis to treat myocardial ischemia via Ang-1/Tie-2/FAK pathway. Front Pharmacol 2022;13:1103557.
143. Ayyadevara S, Mercanti F, Wang X, et al. Age- and Hypertension-associated protein aggregates in mouse heart have similar proteomic profiles. Hypertension 2016;67:1006-13.
144. Erqou S, Thompson A, Di Angelantonio E, et al. Apolipoprotein(a) isoforms and the risk of vascular disease: systematic review of 40 studies involving 58,000 participants. J Am Coll Cardiol 2010;55:2160-7.
145. Hofman A, Ott A, Breteler MM, et al. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer's disease in the Rotterdam Study. Lancet 1997;349:151-4.
146. Zhang Y, Jiang DS, Yan L, Cheng KJ, Bian ZY, Lin GS. HSP75 protects against cardiac hypertrophy and fibrosis. J Cell Biochem 2011;112:1787-94.
147. Meyer GR, De Keulenaer GW, Martinet W. Role of autophagy in heart failure associated with aging. Heart Fail Rev 2010;15:423-30.
148. Ingham RJ, Gish G, Pawson T. The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene 2004;23:1972-84.
149. Yamano K, Yatsukawa Y, Esaki M, Hobbs AE, Jensen RE, Endo T. Tom20 and Tom22 share the common signal recognition pathway in mitochondrial protein import. J Biol Chem 2008;283:3799-807.
150. Alagpulinsa DA, Ayyadevara S, Yaccoby S, Shmookler Reis RJ. A cyclin-dependent kinase inhibitor, dinaciclib, impairs homologous recombination and sensitizes multiple myeloma cells to PARP inhibition. Mol Cancer Ther 2016;15:241-50.
151. Konstantinidis K, Whelan RS, Kitsis RN. Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol 2012;32:1552-62.
152. Thorp EB. The myocardial unfolded protein response during ischemic cardiovascular disease. Biochem Res Int 2012;2012:583170.
153. Iwai T, Tanonaka K, Inoue R, Kasahara S, Kamo N, Takeo S. Mitochondrial damage during ischemia determines post-ischemic contractile dysfunction in perfused rat heart. J Mol Cell Cardiol 2002;34:725-38.
154. Jin JK, Blackwood EA, Azizi K, et al. ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling pathways in the heart. Circ Res 2017;120:862-75.
155. Ganne A, Balasubramaniam M, Mainali N, Atluri P, Shmookler Reis RJ, Ayyadevara S. Physiological consequences of targeting 14-3-3 and its interacting partners in neurodegenerative diseases. Int J Mol Sci 2022;23:15457.
156. Ayyadevara S, Balasubramaniam M, Parcon PA, et al. Proteins that mediate protein aggregation and cytotoxicity distinguish Alzheimer's hippocampus from normal controls. Aging Cell 2016;15:924-39.
157. Bowroju SK, Mainali N, Ayyadevara S, et al. Design and synthesis of novel hybrid 8-hydroxy quinoline-indole derivatives as inhibitors of aβ self-aggregation and metal chelation-induced aβ aggregation. Molecules 2020;25:3610.
158. Balasubramaniam M, Mainali N, Bowroju SK, et al. Structural modeling of GSK3β implicates the inactive (DFG-out) conformation as the target bound by TDZD analogs. Sci Rep 2020;10:18326.
159. Kakraba S, Ayyadevara S, Penthala NR, et al. A novel microtubule-binding drug attenuates and reverses protein aggregation in animal models of alzheimer’s disease. Front Mol Neurosci 2019;12:310.
160. Ganne A, Balasubramaniam M, Griffin WST, Shmookler Reis RJ, Ayyadevara S. Glial fibrillary acidic protein: a biomarker and drug target for alzheimer’s disease. Pharmaceutics 2022;14:1354.
161. Abohassan M, Alshahrani M, Alshahrani MY, Rajagopalan P. In silco and in vitro approaches identify novel dual PI3K/AKT pathway inhibitors to control acute myeloid leukemia cell proliferations. Med Oncol 2022;39:249.
162. Shati AA, Eid RA, Zaki MSA, Alqahtani YA, Al-Qahtani SM, Chandramoorthy HC. Lycopene scavenges cellular ROS, modulates autophagy and improves survival through 7SK snRNA interaction in smooth muscle cells. Cells 2022;11:3617.
163. Burgoyne JR. Oxidative stress impairs autophagy through oxidation of ATG3 and ATG7. Autophagy 2018;14:1092-3.
164. Bhuiyan MS, Pattison JS, Osinska H, et al. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest 2013;123:5284-97.
165. Zhang P, Zhang J, Zhang Y, Wang S, Pang S, Yan B. Functional variants of the ATG7 gene promoter in acute myocardial infarction. Mol Genet Genomic Med 2018;6:1209-19.
166. Balasubramaniam M, Ayyadevara S, Ganne A, et al. Aggregate interactome based on protein cross-linking interfaces predicts drug targets to limit aggregation in neurodegenerative diseases. iScience 2019;20:248-64.
167. Ayyadevara S, Balasubramaniam M, Kakraba S, Alla R, Mehta JL, Shmookler Reis RJ. Aspirin-mediated acetylation protects against multiple neurodegenerative pathologies by impeding protein aggregation. Antioxid Redox Signal 2017;27:1383-96.
168. Ayyadevara S, Balasubramaniam M, Suri P, et al. Proteins that accumulate with age in human skeletal-muscle aggregates contribute to declines in muscle mass and function in Caenorhabditis elegans. Aging 2016;8:3486-97.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Mainali N, Ayyadevara S, Ganne A, Shmookler Reis RJ, Mehta JL. Protein homeostasis in the aged and diseased heart. J Cardiovasc Aging 2023;3:16. http://dx.doi.org/10.20517/jca.2023.4
AMA Style
Mainali N, Ayyadevara S, Ganne A, Shmookler Reis RJ, Mehta JL. Protein homeostasis in the aged and diseased heart. The Journal of Cardiovascular Aging. 2023; 3(2): 16. http://dx.doi.org/10.20517/jca.2023.4
Chicago/Turabian Style
Mainali, Nirjal, Srinivas Ayyadevara, Akshatha Ganne, Robert J. Shmookler Reis, Jawahar L. Mehta. 2023. "Protein homeostasis in the aged and diseased heart" The Journal of Cardiovascular Aging. 3, no.2: 16. http://dx.doi.org/10.20517/jca.2023.4
ACS Style
Mainali, N.; Ayyadevara S.; Ganne A.; Shmookler Reis RJ.; Mehta JL. Protein homeostasis in the aged and diseased heart. J. Cardiovasc. Aging. 2023, 3, 16. http://dx.doi.org/10.20517/jca.2023.4
About This Article
Copyright

Data & Comments
Data


 Cite This Article 16 clicks
Cite This Article 16 clicks

 Like This Article 32
likes
Like This Article 32
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.