
Mitochondrial DAMPs-dependent inflammasome activation during aging induces vascular smooth muscle cell dysfunction and aortic stiffness in low aerobic capacity rats


Abstract
Introduction: Low aerobic exercise capacity is an independent risk factor for cardiovascular disease (CVD) and a predictor of premature death. In combination with aging, low aerobic capacity lowers the threshold for CVD.
Aim: Since low aerobic capacity and aging have been linked to mitochondrial oxidative stress and dysfunction, we investigated whether aged Low-Capacity Runner (LCR) rats (27 months) had vascular dysfunction compared to High-Capacity Runner (HCR) rats.
Methods and Results: A significant decrease in aortic eNOS levels and vasodilation as well as an increase in aortic collagen and stiffness were observed in aged LCR rats compared to age and sex-matched HCR rats. There was a correlation between age-related vascular dysfunction and increased levels of ROS and DNA damage in aortas of LCR rats. Moreover, mitochondrial oxygen consumption, membrane potential, ATP levels, and mitophagy were lower in VSMCs of aged LCR rats. VSMCs from older LCR rats showed AIM2 inflammasome activation. VSMCs of young (4 months old) LCR rats treated with purified mitochondrial damage-associated molecular patterns (DAMP) recapitulated an inflammasome activation phenotype similar to that seen in aged rat VSMCs. Rapamycin, a potent immunosuppressant, induced mitophagy, stimulated electron transport chain activity, reduced inflammasome activity, mitochondrial ROS and DAMP levels in VSMCs from aged LCR rats. MitoTEMPO, a mitochondrial ROS scavenger, was similarly effective on VSMCs from aged rats.
Conclusion: The findings suggest that impaired mitophagy and inflammasome activation in the vasculature under conditions of low aerobic exercise capacity during aging results in arterial dysfunction and aortic stiffness. In older adults with reduced aerobic capacity, mitochondrial antioxidants, mitophagy induction, and inflammasome inhibition may be effective therapeutic strategies for enhancing vascular health.
Keywords
INTRODUCTION
Both aerobic exercise capacity and activity are well-established predictors of age-adjusted cardiovascular and overall mortality in both healthy subjects and those with cardiovascular disease (CVD)[1-5]. Peak aerobic exercise capacity declines with age, and the rate of decline increases markedly with each successive decade[6]. A decrease in the peak heart rate, peripheral oxygen consumption, cardiac β-adrenergic receptor density, and postsynaptic β-adrenergic signaling underlies the decline in aerobic capacity associated with aging, which lowers the threshold for CVD development.
Most of the population studies on exercise capacity focus on correlation-based assessment of patients without attempting to infer a causal relationship between the variables. Experimental validation of causal inference, if any, in correlation-based population studies was performed using unique rat models with high and low intrinsic aerobic capacities (High-Capacity Runners, HCR and Low-Capacity Runners, LCR)[7]. Specifically, this was accomplished by genetic segregation via two-way artificial selection based on treadmill exercise tests to investigate the influence of intrinsic aerobic capacity on all-cause mortality and morbidity[8]. The median lifespan for LCR is 28%-45% shorter than that for HCR, and maximal oxygen uptake serves as a reliable predictor of lifespan across adulthood[9]. In addition, for every 10 g gain in body weight during adulthood, lifespan decreased by approximately 1 month in a combined population of male and female rats, indicating that gain in body weight over time is also an important predictor of longevity. Early deaths in LCR may be attributed to pathophysiological mechanisms associated with aging, such as the failure of one or more organs. The LCR rats also exhibit a number of cardiovascular risk factors and impaired mitochondrial oxidative pathways[10].
The impaired mitochondrial oxidative phosphorylation increases mitochondrial reactive oxygen species (ROS) production, resulting in mitochondrial DNA (mtDNA) damage[11,12]. Mitochondrial ROS, in turn, impair mitochondrial respiration, limiting exercise capacity[13,14]. The increased production of mitochondrial ROS, accumulation of mtDNA damage, and progressive respiratory dysfunction contribute to the pathogenesis of CVD[12,15], and a causative link has been established between DNA damage, both nuclear and mitochondrial, and aging-related phenotypes[16-20]. Mitophagy, the selective degradation of mitochondria by autophagy[21], removes the damaged mitochondria to maintain cellular homeostasis[22]. Impairment of mitophagy results in diminished clearance of damaged mitochondria, which releases mtDNA into circulation[23-26]. Known as damage-associated molecular patterns (DAMPs), mitochondria-derived fragments are implicated in the development of chronic (sterile) inflammation in aging[25] via activation of inflammasome[27]. Additionally, mitochondrial dysfunction and chronic systemic inflammation have been linked to obesity[28,29].
The hardening of the central arteries is a hallmark of advancing age, which impacts cardiac function, microcirculation, and organ function[30,31] and is a major cause of age-related CVD[32]. Lower cardiorespiratory fitness is correlated with arterial stiffening in coronary artery disease patients with myocardial infarction[33], and higher cardiorespiratory fitness is correlated with a reduction in age-related arterial stiffness in adults[34]. There is also evidence that obesity accelerates the process of artery stiffening earlier in life[35,36]. It has been proposed that defective mitochondrial quality control contributes to the thickening of large elastic arteries and stiffening with age via mitochondrial stress/dysfunction[30]. However, the molecular mechanisms regulating aortic stiffening are yet to be fully elucidated.
This study examines VSMC function and metabolism in aging at the intersection of low aerobic capacity and obesity phenotype. We hypothesized that reduced aerobic capability in older LCR rats would be associated with impaired vascular function, increased aortic stiffening, elevated mitochondrial oxidative stress, mtDNA damage, impaired mitophagy, and inflammasome activation, along with vascular remodeling.
MATERIALS AND METHODS
Rats: All animal procedures were performed in compliance with the protocols approved by the Institutional Animal Care and Use Committee of the University of Michigan (PRO00010178) in accordance with NIH OLAW guidelines. The Low-Capacity Runner (LCR) and High-Capacity Runner (HCR) rat models have been described previously[7]. Exercise capacity was the basis for artificial selection in a genetically heterogeneous population of rats (N:NIH outcrossed stock) to isolate LCR and HCR rats. At 11 weeks of age, every rat underwent endurance running tests on a treadmill, and the time and maximal distance it was able to run before exhaustion were monitored. The top 20 percent and the bottom 20 percent of the runners were bred for genetic heterogeneity for 40 generations in order to segregate LCR and HCR rats[9]. We used young
RNASeq: RNA was extracted from thoracic aortas of young and old HCR and LCR rats (n = 3) using the Qiagen RNeasy Kit. RNA quality was assessed using an Agilent 2100 Bioanalyzer. All RNA samples had an RNA Integrity Number of ≥ 9.5. Sequencing libraries were prepared at University of Michigan Advanced Genomics Core with TruSeq RNA Library Preparation Kit (Illumina). RNA sequencing was performed on an Illumina HiSeq 4000 platform with three lanes per sample generating 125 bp paired-end reads with an average of 80 million reads per sample. Quality control and alignment for RNA sequencing were performed at the University of Michigan Bioinformatics Core. Raw reads were concatenated into a single FASTQ file for each sample. The quality of the raw reads was checked with FastQC. Reads were aligned to the reference genome (UCSC rn5.fa) using Tuxedo Suite: TopHat and Bowtie2[37]. Cufflinks was used for post-alignment quality control, expression quantification, and normalization. iDEP.91 was used to perform hierarchical clustering, principal component analysis, and differential gene expression analysis with default parameters (Available from: http://bioinformatics.sdstate.edu/idep/)[38]. We identified differentially expressed transcripts and genes using the FDR ≤ 0.05 and fold change ≥ ±1.5. Differentially expressed genes were annotated using gene ontology terms, and pathway enrichment analyses were performed using the g:Profiler g:GOSt web application (Available from: http://biit.cs.ut.ee/gprofiler/gost)[39]. To identify enriched signaling pathways, we used the KEGG Pathway database (Available from: https://www.genome.jp/kegg/pathway.html).
VSMC culture: Four separate VSMC isolates were made from the aortas of young (4-month-old) and old (27-month-old) HCR and LCR rats. Details of the cell isolation protocol were published previously[40]. Briefly, aortic VSMCs were isolated by stripping off the adventitial layer and using only the media for cell isolation. The adventitial and medial layer separation was confirmed by immunostaining with α-SM actin antibody of random samples. Before digestion of the media to obtain VSMCs, calcium alginate swabs were used to remove the endothelium. Aortic rings (< 3 millimeters) were then incubated in collagenase type II and elastase mixture at 37 °C for 1.5 h or until complete digestion with intermittent mixing with a glass Pasteur pipet. Released VSMCs were cultured in DMEM supplemented with 10% FBS, antibiotics, and Amphotericin B (Gibco). Experiments were performed with cells from passages 3-10. Gene expression analysis of Acta2 (Actin Alpha 2, Smooth Muscle), Myh11 (Myosin heavy chain 11), and Cnn2 (Calponin 2) confirmed the phenotype of VSMCs in all isolates.
ROS detection: Incubation of frozen aortic sections or cultured VSMCs in Hank’s balanced salt solution containing 1 mM CaCl2 and 1 mM MgSO4 for 15 min was performed in the presence of
Immunofluorescence: For Immunofluorescent staining, the tissues or cells were fixed in 3.6% formaldehyde for 20 min at room temperature and permeabilized in 0.05% Triton X-100 for 3 min, followed by 3 × 5 min washings in PBS. The fixed tissues or cells were incubated with primary antibodies [Supplementary Table 1] overnight at 4 °C according to the manufacturer’s instructions. Thereafter, the tissue was washed three times with PBS and incubated with an Alexa Fluor-conjugated secondary antibody for 2 h. The Auto-fluorescence was quenched with TrueVIEW kit (Vector laboratories). The slides were mounted in mounting media containing DAPI (H100, Vector labs).
Picrosirius red staining: The aortic sections were fixed in acetone and cold methanol for 30 min each. Sections were stained with Picrosirius red kit (Abcam). The excess stain was washed off twice with acetic acid, and the slides were dehydrated with ethanol. Sections were mounted using Vectashield H100 mounting medium without DAPI. The images were acquired under brightfield using a Nikon TI-E camera attached to an upright E600 microscope at equal exposure time for all images. NIH Image J was used to measure the area of picrosirius red staining in the aortic media.
Lipofuscin staining: The VSMCs were cultured in 8-well chambered glass slides until 80% confluency was reached. Cells were washed twice with PBS at room temperature, then incubated for 3 min in cold Glutaraldehyde fixative solution (25 mL acetone and 75 mL glutaraldehyde solution) for 3 min and washed thoroughly with PBS. Next, cells were stained with Sudan Black B staining reagent (Sigma Aldrich) for
Mitophagy: VSMCs were cultured in 8-well chambered slides until 60% confluency was reached. Cells were incubated for 30 min at 37 °C with Mtphagy dye (Mitophagy kit, Dojindo), followed by mitophagy-inducing compounds or vehicle for 16 h. The cells were washed twice with PBS and then exposed to Lyso dye (Mitophagy kit, Dojindo) for another 30 min at 37 °C, according to the manufacturer’s protocol. The slides were washed twice with 1xPBS and mounted in mounting media containing DAPI (H100, Vector labs). All slides were imaged by Nikon Eclipse Ti- fusion microscope with DAPI, FITC and Texas Red Channels at equal exposure, gain and offset. The colocalization of mtphagy and lyso dyes (yellow color) was quantified by NIH Image J as integrated density per area of interest. An indicator of mitophagy flux is the ratio of colocalization signals in induced samples to those in uninduced samples. We then measured autophagic vacuole formation using the CYTO-ID kit (ENZ-KIT175-0050, ENZO) according to the manufacturer’s protocol. Briefly, the VSMCs were cultured until 80% confluency in 8-well chambered slides. The attached cells were incubated with the working solution containing CYTO-ID dye for 30 min at 37 °C. This was followed by two washes with 1xPBS and mounting in mounting media containing DAPI (H100, Vector labs) and imaging.
Flow cytometry: The cells were detached by trypsinization, then placed in a 70 µM cell strainer (Corning, Corning, NY) and washed with FACS buffer (PBS, 1% BSA, 1 mM EDTA). The cells were stained with Apoptosis, DNA Damage and Cell Proliferation Kit (562253 BD Biosciences, Franklin Lakes, NJ) following the manufacturer’s instructions. The cells were processed using the ZE5 cellular analyzer (Bio-Rad, Hercules, CA) and the data were analyzed with FlowJo v10 (FlowJo, LLC, Stanford University, CA).
Measurement of ATP levels: Cellular ATP levels in both young and old HCR and LCR VSMCs were measured using a bioluminescence ATP determination kit (ThermoFisher), following the manufacturer’s instructions. In this assay, ATP reacts with luciferin in the presence of luciferase, converting it into oxyluciferin. As a result, the light produced is proportional to the cellular ATP levels[41]. In brief, equal volumes of VSMC protein lysates were incubated with a reaction buffer containing D-luciferin (500 M), firefly luciferase (1.25 g/mL), and 1 mM (DTT). The luminescence was measured with a luminometer, and ATP levels were determined using a standard curve generated simultaneously.
Western blotting: VSMC and aortic protein lysates were prepared using M-PER and T-PER lysis buffers, respectively, and the protein concentration was determined using BCA assay (ThermoFisher). Equal amounts of proteins were loaded on SDS-PAGE gels and were separated by electrophoresis. The proteins were then transferred onto nitrocellulose membranes using a semi-dry Western transfer apparatus (Trans-Blot Turbo system, BioRad). The blots were blocked with BlockerTM BSA (ThermoFisher) and probed overnight with primary antibodies (AIM2, Caspase1, VCAM, LAMP1 from Santa Cruz; IL18, TOM20 from Abcam; FITC- tagged-ACTA2 from BIOS). Blots were washed 3 × 5 min with PBST
Mitochondrial bioenergetics: The mitochondrial bioenergetics of rat VSMCs were measured using the Seahorse XFp and XF96 machines (Agilent technologies). Approximately 20,000 cells were cultured in DMEM supplemented with 10% FBS and antibiotics in cell culture microplates supplied with Flexpack (Agilent technologies) for 24 h. The next day, the media were changed to phenol-red free basal DMEM (Agilent technologies) supplemented with 25 mM glucose, 2 mM pyruvate, and 1 mM glutamine, and incubated in a CO2-free incubator at 37 °C for least 1 h before analysis. Cellular oxygen consumption rate (OCR) at basal level and in the presence of 2 M oligomycin, 500 nM trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP) and 500 nM rotenone + antimycin A was quantified in three consecutive cycles for 3 min each. Pre-treatment of cells with 20 nM rapamycin or 10 nM MitoTEMPO were performed where indicated for 16-18 h. In order to normalize the OCR values to protein levels, the cells were lysed with M-PER buffer and the protein content was determined by BCA assay.
Mitochondrial DNA damage analysis: The total genomic DNA of rat aortas was isolated with a Qiagen blood and tissue kit. The DNA concentration was determined using two different methods, the Nano-Drop ND-1000 spectrophotometer and the Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific). Equal amounts of DNA were used as a template to amplify a 13.4 kb mitochondrial DNA fragment. We used rat-specific primers (Forward: AAA ATC CCC GCA AAC AAT GAC CAC CC, Reverse: GGC AAT TAA GAG TGG GAT GGA GCC AA)[42] and Takara LA TaqTM long DNA polymerase (Takara Bio USA) for amplification of mitochondrial DNA in a polymerase chain reaction. An equal aliquot of the PCR product was run on an agarose gel to determine the quality and densitometry to determine the quantity of the expected size band. The amount of amplification product from the long-fragment PCR was normalized by measuring the levels of MTCO1 in the DNA template in a separate quantitative PCR reaction using a Taqman probe.
Cell-free DNA isolation and amplification: An equal number of VSMCs from young and old LCR and HCR rats were plated in 6-well culture dishes. After 30 h of incubation, the media were spun down at
DAMPs preparation: MtDNA was isolated from rat liver mitochondria using DNA blood and tissue kit (Qiagen). Firstly, mitochondria were isolated with mitochondrial isolation kit (Pierce), and then mitochondrial pellets were used to isolate DNA. DNA was sonicated up to 5 cycles on ice, at a low setting, each for 30 s, spaced by 1-min intervals. The DNA samples before and after the sonication were run on 1.5% agarose gel to determine the size of the fragmented DNA samples. A length of 150 to 250 base pairs was considered optimal to induce inflammasome activation in cells.
Wire myography: The vascular function analysis was performed using a Danish Myo Technology wire myograph (DMT 620 MS, ADInstruments, Inc., Colorado Springs, CO) as described[16]. In brief, the mesenteric arteries were freshly isolated and immediately mounted using 25 µm tungsten wires to the jaws of the DMT myograph. The vessels were kept in physiological saline solution (PSS; 130 mM NaCl, 4.7 mM KCl, 1.18 mM KH2PO4, 1.17 mM MgSO4·7H2O, 14.9 mM NaHCO3, 5.5 mM dextrose,
Pressure myography: The descending thoracic aorta segments were isolated from LCR and HCR rats
Statistics: All biochemical experiments were repeated at least three times. All analyses were performed with Prism 8 (GraphPad Software). All data were examined for normality using the Shapiro-Wilk test. Where applicable, data were analyzed using an unpaired 2-tailed t-test. For experiments involving more than two groups, one-way ANOVA with Tukey post-hoc tests were used. Differences were considered significant at
RESULTS
The interplay between aerobic exercise capacity and aging affects arterial function, inducing aortic stiffening
The development of aortic stiffening is a key feature of aging and an independent predictor of cardiovascular events in hypertensive patients as well as in the general population[45-47]. In this study, we investigated the interaction between exercise capacity, aging, and arterial stiffness. First, we investigated the vasomotor function of mesenteric artery rings by wire myography; however, it did not indicate any significant differences in phenylephrine-induced maximum contraction between old age LCR and HCR rats [Figure 1A]. We then measured acetylcholine (ACH)-induced relaxation of mesenteric arteries from young and old age rats. The relaxation response to ACH was similar in young LCR and HCR rats [Figure 1B]. However, relaxation of the mesenteric arteries from old age LCR rats was significantly smaller than that from old age HCR rats at lower concentrations of ACH, with a marked rightward shift in the concentration-response curve [Figure 1C] and a smaller half-maximal relaxation response [-log(EC50); Figure 1D].
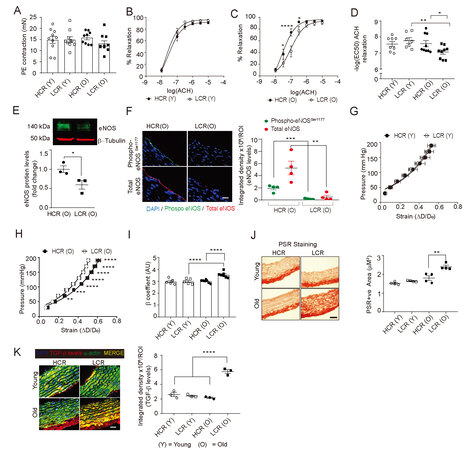
Figure 1. Increased vascular dysfunction, fibrosis, and stiffness in LCR rats with aging. The contractile force in mesenteric artery rings of young (4-month old) and aged (27-month old) LCR and HCR rats in response to phenylephrine (PE) was measured by wire myography (mean ± SEM; n = 5) (A). Concentration-response curves for acetylcholine (ACH) induced relaxation of mesenteric arteries from young (B) and old age (C) LCR and HCR rats (mean ± SEM; n = 5). Half-maximum relaxation concentration of ACH [-log(EC50)] was calculated from independent dose-response curves and plotted as mean ± SEM, n = 5 (D). Representative Western blot of eNOS protein expression in aortic lysates. Densitometry analysis of eNOS protein levels normalized to β-tubulin levels (mean ± SEM, n = 3) (E). Representative fluorescence images of aortic cross sections stained for immunoreactive total eNOS (red) or phospho-eNOS Ser1177 (green). Data are presented as mean integrated density/ROI ± SEM where n = 4 [(F), scale = 50 μm)]. Pressure vs strain data of thoracic aorta segments from young (G) and old (H) LCR and HCR rats were analyzed using pressure myography. The relative stiffness coefficient (H) was calculated by fitting pressure vs strain data to an exponential formula (mean ± SEM, n = 5) (I). Representative images of rat aortic cross sections stained with Picrosirius red (PSR) (J). Representative fluorescence images of aortic cross sections co-stained for TGF-β (red) and SM-α-actin (green). Bright yellow fluorescence indicates colocalization of TGF-β and SM-α-actin. Data are presented as mean integrated density/ROI ± SEM where n = 4 (K). *P < 0.05; **P < 0.01; ****P < 0.0001.
We asked whether a decrease in eNOS expression might be responsible for arterial dysfunction in old LCR rats, since nitric oxide derived from eNOS is important for vascular function. The Western blot analysis of aortic lysates of old age LCR rats revealed a 40% reduction in eNOS protein expression in comparison with the old HCR rats [Figure 1E]. In order to find out if attenuated eNOS phosphorylation and inhibition of its activity contribute to arterial stiffening with low aerobic capacity and aging[48], we analyzed the expression of phospho-eNOS (Ser1177) in the aortas of old LCR and HCR rats. The phospho-eNOS levels in old HCR rats were higher than in old LCR rats [Figure 1F], suggesting that endothelial dysfunction in mesenteric arteries and aortas of LCR rats with aging may be due to reduced eNOS protein expression and activity.
To determine the effects of low aerobic capacity on the function of large conduit arteries during aging, we measured the aortic stiffness of thoracic aortic segments using pressure myography. There was no difference in aortic plasticity between young LCR and HCR rats [Figure 1G]. In contrast, the old LCR rats showed a significant increase in aortic stiffness compared to age-matched HCR rats [Figure 1H and I, P = 0.0002]. The remodeling of the extracellular matrix is a major contributor to vessel wall stiffness in aging, and collagen deposition and cross-linking are key to this process[16,49,50]. We stained fresh frozen aortic cross sections with picrosirius red to measure the collagen content. Aortic collagen levels were not different in young LCR and HCR rats [Figure 1J]. Conversely, old LCR aortas have a 34% higher collagen content than old HCR aortas
Aging is characterized by increased inflammation and necroptosis pathways in the aortic transcriptome of LCR rats
In order to examine how aging and low aerobic capacity impact vascular phenotype at the global level, we performed genome-wide RNA sequencing analysis and compared the transcriptional profiles and signaling pathways enriched in the aortas of young and old HCR and LCR rats. In a hierarchical cluster analysis of the top 1000 most variable genes, there was a distinct separation between young and aged rats, as well as between HCR and LCR rat aortas [Figure 2A]. A principal component analysis showed that old LCR aortas have different transcription profiles from old HCR aortas, besides the obvious differences between young and old rat aortas [Figure 2B]. In the comparison of significantly differentially expressed genes (FDR ≥ 0.05 and fold change ± 1.5) among all groups, the largest differences were found between old HCR and old LCR aortas [Figure 2C]. Transcription analysis revealed a unique expression pattern between old LCR and old HCR aortas with over 1100 genes differentially expressed [Figure 2D].
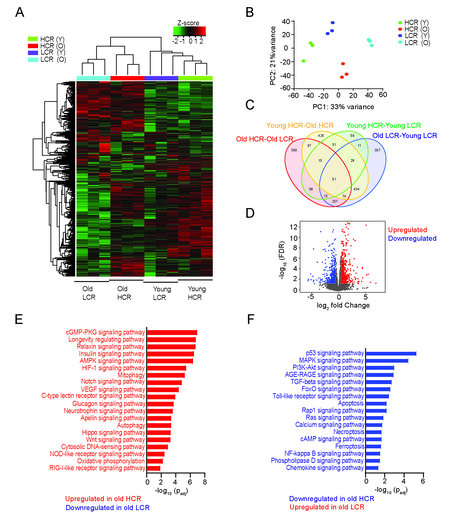
Figure 2. Genome-wide RNA sequencing analysis of aortas from young and old HCR and LCR rats. (A) Heat map representation of unsupervised hierarchical clustering analysis with top 1000 most variable genes. (B) Principal component analysis of the whole transcriptome from young and old HCR and LCR aortas. (C) Venn diagram of differentially expressed genes across four comparisons between young and old HCR and LCR aortas. (D) Volcano plot of differentially expressed genes between old HCR and LCR with FDR ≥ 0.05 and fold change ±1.5. Gene ontology pathway enrichment analysis showing top selected KEGG signaling pathways upregulated (E) or downregulated (F) in old HCR compared with old LCR aortas.
Compared to old LCR aortas, HCR aortas had 555 upregulated genes and 560 downregulated genes. By contrast, the differentially expressed genes in young HCR aortas were 168 upregulated and 160 downregulated compared with those in young LCR aortas. A gene ontology pathway enrichment analysis focused on selected signaling pathways defined in the KEGG database was conducted to better understand phenotypic changes defined by transcriptional profiles. Significantly differentially upregulated transcripts in old HCR aortas were related to longevity regulating pathways, growth factor signaling pathways (insulin, AMPK, glucagon, and neurotrophin signaling), oxidative phosphorylation, DNA/RNA sensing (RIG-I receptor signaling), and mitophagy/autophagy pathways[Figure 2E]. In contrast, genes significantly upregulated in old LCR aortas were associated with cell cycle regulation (p53 and FoxO signaling) and pro-inflammatory pathways (AGE-RAGE, TGF-β, Toll-like receptor, NF-kappa B, and chemokine signaling) and apoptosis/necroptosis/ferroptosis regulating pathways [Figure 2F]. Based on pathway enrichment analysis, old LCR aortas have reduced mitochondrial biogenesis and mitophagy, increased damage and cytosolic accumulation of DNA/RNA, pro-inflammatory pathway activation, and enhanced apoptosis and necroptosis.
Low aerobic capacity in old age is associated with increased mitochondrial oxidative stress, mitochondrial DNA (mtDNA) damage and dysfunction
We and others have demonstrated that mitochondria-derived oxidative stress increases with aging and affects vascular function and arterial stiffness in animal models and humans[16,40,53-55]. VSMC plasticity plays a pivotal role in vascular remodeling in aging[56,57], which is why we investigated many mechanisms of mitochondrial dysfunction in both the aorta and VSMCs to determine how low aerobic exercise capacity impacts VSMC function. VSMCs isolated from 4-month-old rats did not show any differences in proliferation or senescence between LCRs and HCRs. Aortic ROS levels and mitochondrial ROS levels (mtROS) were measured in aortas as well as in the aortic VSMCs of young and old LCR and HCR rats. Aortic ROS levels measured by DHE fluorescence were not significantly different between both young and old age LCR and HCR rats [Supplementary Figure 1]. However, a higher percentage of VSMCs from old LCR rats had comet tails than VSMCs from old HCR rats, indicating increased nuclear DNA damage
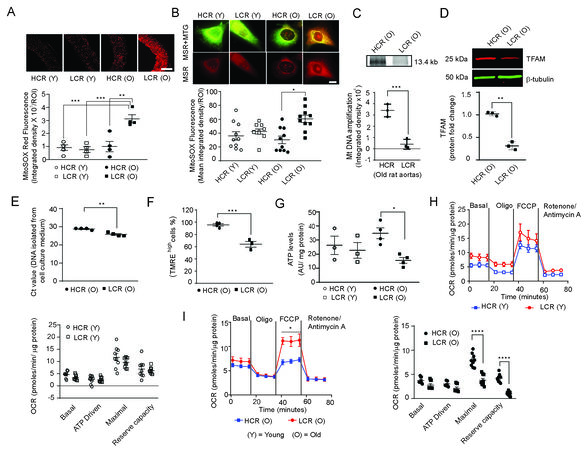
Figure 3. LCR rats show increased mitochondrial oxidative stress, mtDNA damage, circulating mitochondrial DAMPs and dysfunction in old age. Representative fluorescence images of aortic cross sections stained with MitoSOX Red (MSR) (A, top panel, scale 50 μm) and VSMCs stained with MitoTracker Green (MTG) and MitoSOX Red (B, top panel, scale 10 μm) from young and old age HCR and LCR rats. MitoSOX Red fluorescence was presented as integrated density (mean ± SEM, n = 4) (A and B bottom panels). Representative long PCR amplicon of 13.4 kb mitochondrial DNA from aortas (C, top panel) from old age HCR and LCR rats. PCR product band intensities were normalized to a short amplicon of 0.22 kb GAPDH (mean ± SEM, n = 3) (C, bottom panel). Western blot analysis and densitometric quantification of TFAM in VSMC lysates (D, top panel). Data are presented as fold change ± SEM where n = 4
Since increased mitochondrial oxidative stress can cause mtDNA damage[58,59], we investigated mtDNA integrity by long PCR amplification of a 13.4 kb mtDNA fragment. Consistent with the increase in mtROS levels, an 8.6-fold lower mtDNA amplification was observed in aortas from aged LCR rats compared to aged HCR rats [Figure 3C]. We wondered if this dramatic reduction in mtDNA amplification was due to an additive effect of mitochondrial ROS and the loss of protective mechanisms. Western analysis for TFAM in the VSMC lysates showed a 3.3-fold decrease in old LCRs compared to old HCRs [Figure 3D]. Based on these results, low aerobic capacity-induced mtROS contribute to mtDNA damage in old age, impairing the replication of mtDNA.
Because aging can lead to increased production of damage-associated molecular patterns (DAMPs)[25], we investigated the effect of the interaction of low aerobic capacity and aging on mtDNA damage as measured by QPCR with a short Taqman probe specific for MTCO1 gene amplification in VSMC culture medium of aged LCR and HCR rats. The Ct value for MTCO1 amplification decreased significantly in DNA amplified from VSMC culture media from old age LCR rats compared with that in HCR rats, which is inversely related to the amount of extracellular MTCO1 DNA present in the culture media [> 2 Ct value decrease,
Because extrusion of cellular mtDNA occurs following mitochondrial depolarization[23], we measured mitochondrial membrane potential in rat aortic VSMCs from old age LCR and HCR rats by flow cytometric analysis of TMRE retention[60]. A 30% decrease in TMREhigh cells was observed in old age LCR rat VSMCs compared with HCR rat cells [Figure 3F]. Mitochondrial membrane potential harnesses proton motive force to generate ATP. A colorimetric assay was used to compare levels of ATP in VSMCs from young and old age LCR and HCR rats. The VSMCs from older LCR rats exhibited 2.24-fold lower ATP levels (P < 0.05) than VMSCs from younger HCR rats, while cells from young LCR and HCR rats exhibited equal ATP levels [Figure 3G].
The mtDNA-encoded proteins are essential components of respiratory complex subunits, and mtDNA damage compromises cellular respiration[61]. We measured oxygen consumption rate (OCR) to examine the effects of low aerobic exercise capacity as well as aging on VSMC respiration. The OCR of VSMCs from young LCR and HCR rats was not significantly different [Figure 3H]. However, the maximal OCR by complex IV in the presence of FCCP, which collapses the proton gradient and disrupts the mitochondrial membrane potential and enables an uninhibited electron transport chain, was significantly lower in VSMCs from old age LCR compared to HCR rats [Figure 3I]. In addition, the reserve respiratory capacity, defined as the difference between maximal and basal respiration, was significantly lower in VSMCs from old age LCR versus HCR rats [Figure 3I]. Together, these data confirm that low aerobic capacity negatively affects the ability of conduit arteries in LCR rats to respond to increased energy demands or stresses, particularly as the animals age.
LCR rats exhibit impaired mitophagy in aging
Inhibition of mitophagy, the autophagy of mitochondria, causes the accumulation of dysfunctional mitochondria and extrusion of mitochondrial components[25]. As we observed increased mitochondrial damage and higher levels of mtDNA DAMPs, we investigated whether LCR rats suffer from impaired mitophagy in old age. Mitophagy was measured as integrated density from the colocalization of immunofluorescent red mitochondrial marker protein TOM20 with green lysosomal marker protein LAMP1 and blue SM α-actin in aortas and from the colocalization of red TOM20 and green LAMP1 in VSMCs. Older LCR rats displayed a 54% decrease in mitophagy in medial SMCs of aortas [Figure 4A], which was also supported by a 76% reduction in cultured VSMCs [Figure 4B; Supplementary Figure 4] in comparison to older HCR rats. The mitophagy levels did not differ significantly between young age LCR and HCR rats.
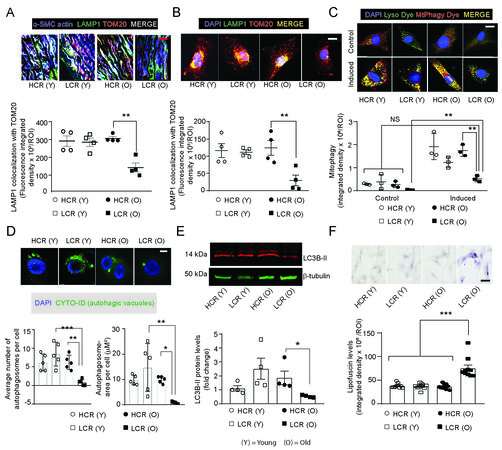
Figure 4. VSMCs of the aortic media and cultured aortic VSMCs of old age LCR rats show impaired mitophagy. Representative confocal images of aortic cross sections (A, top panel) were co-stained for LAMP1 (green), TOM20 (red) and SM-α-actin (blue). The white fluorescence indicates colocalization of all proteins and active mitophagy in medial VSMCs. VSMCs (B, top panel) were co-stained for LAMP1 (green) and TOM20 (red). Bright yellow fluorescence indicates colocalization of LAMP1 and TOM20 and active mitophagy. Data are presented as mean integrated density/ROI ± SEM where n = 4 independent cell lines (A and B, bottom panels). Representative confocal images of mitophagy flux, showing bright yellow colocalization of Mtphagy-(red) and Lyso-(green) dyes (C, top panel; scale
Mitophagy induction in live VMSCs was observed using Mtphagy Dye, which emits a high signal when damaged mitochondria fuse with lysosomes (Mitophagy kit, Dojindo) in the presence of rapamycin, a well-known mitophagy inducer[62]. The fusion of Mtphagy Dye-labeled mitochondria and lysosomes was confirmed with Lyso Dye. The rapamycin-induced mitophagy in VSMCs of both young and old LCR and HCR rats [Figure 4C]. However, mitophagy induction was higher in VSMCs of young HCR rats than in old LCR rats, and in VSMCs of old HCR rats than in old LCR rats.
The autophagic vacuole formation is one of the essential steps in transporting damaged mitochondria into lysosomal compartments. We measured the formation of autophagic vacuoles in VSMCs by measuring the green-fluorescent puncta that form when CYTO-ID (ENZO Inc.) is specifically localized to autophagic vacuoles. In old LCR VSMCs, green fluorescence area of puncta was 12.0-fold lower than in old HCR VSMCs, indicating impaired autophagy flux, but this difference was not found in young HCR and LCR VSMCs [Figure 4D]. Western blot analysis of VSMC lysates also revealed a decreased level of LC3B-II, a marker of autophagosomes, in old LCR rats compared to old HCR rats [Figure 4E, P < 0.05]. Additionally, we tested the effects of autophagic flux inhibitor Bafilomycin A1 (50 nM for 16 h) on LC3B-II and autophagic adapter p62 levels in VSMCs. The levels of p62 [Supplementary Figure 5A] and LC3B-II
Inflammasome activation in aging is associated with diminished aerobic capacity
The inhibition of mitophagy activates inflammasomes, cytosolic danger sensing platforms via increased mitochondrial ROS generation and DAMPs[27,63]. Damaged mtDNA is actively extruded from cells via exosomes, and cytosolic DNA activates the AIM2 inflammasome[64]. Hence, we investigated whether impaired mitophagy and elevated DAMP levels in old LCR rat VSMCs increase the expression of AIM2, caspase-1 and IL-18, which are downstream effectors of AIM2 inflammasome activation. As shown in Figure 5A, immunofluorescence studies indicated a marked increase in AIM2
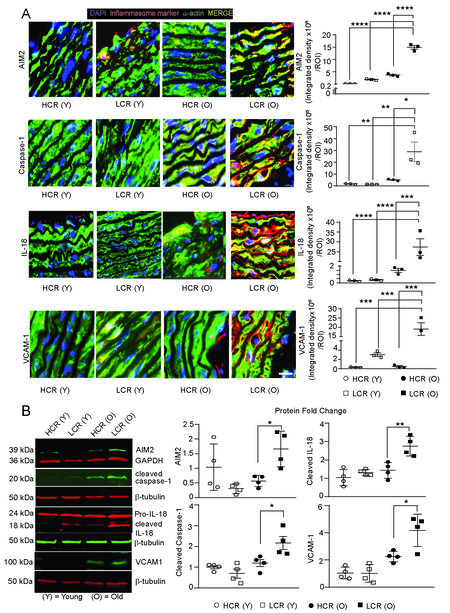
Figure 5. Inflammasome is activated in aortic medial VSMCs and in cultured aortic VSMCs of old age LCR rats. Representative confocal images of aortic cross sections stained for inflammasome activation markers (AIM2, caspase-1, IL-18, and VCAM1) in young and old age rats (A, left panels, red). Aortas were co-stained for ACTA2 (green). The bright yellow fluorescence indicates activation of specific inflammasome markers in medial VSMCs. Data are presented as average integrated density/ROI ± SEM where n = 4 (A, right panels). Representative Western blots showing expression of the inflammasome markers in VSMC lysates (B, left panel). Data are presented as fold change, mean ± SEM, n = 4 (B). Scale is 50 μm. *P < 0.05; **P < 0.01; ***P < 0.001.
In consonance with inflammasome activation, VCAM-1, a major regulator of leukocyte adhesion and transendothelial migration that is upregulated in chronic inflammatory conditions[65], also increased by
Rapamycin and MitoTEMPO attenuate inflammasome activation in aortic VSMCs from old age LCR rats
To determine whether mtDNA damage is sufficient to induce inflammasome expression and activation, VSMCs from young LCR rats were treated with liver mtDNA DAMPs at a concentration of 10 ng/mL for
Since rapamycin inhibits AIM2 activation[66] and reduces aortic stiffness with aging[67], we investigated whether rapamycin inhibits AIM2 pathway activation in aged LCR rat VSMCs. Rapamycin decreased cleaved caspase-1 and cleaved IL-18 levels by 2 and 7.5-fold, respectively [Figure 6C and D,
Next, we tested whether inhibition of inflammasome activation resulted from decreased mtDNA DAMP generation. From rapamycin-treated and untreated VSMC culture media, cell-free DNA was isolated and mtDNA DAMP levels were determined by analyzing the MTCO1 gene by quantitative PCR. The rapamycin treatment reduced mtDNA amplification by 4.5 ± 1.7 cycles (P < 0.05) [Figure 6G]. MitoTEMPO also reduced mtDNA DAMP levels in the culture medium of old age LCR rats [Figure 6G], supporting the notion that inflammasome activation increases mtDNA DAMPS levels, exacerbating inflammation in a feedforward manner.
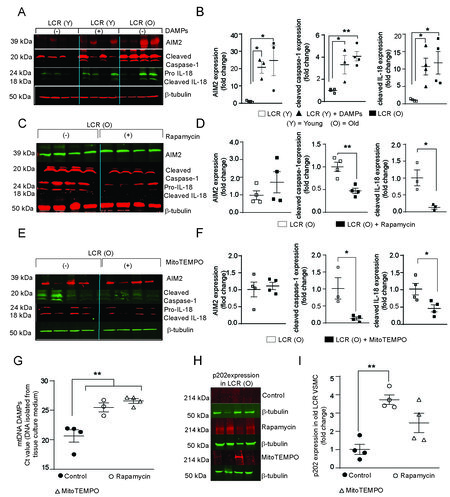
Figure 6. Rapamycin or MitoTEMPO treatment inhibited inflammasome activation in VSMCs from old LCR rats. Representative Western blots of inflammasome marker proteins of VSMCs treated with or without DAMPs (A). Densitometric quantification of proteins expressed as fold change ± SEM, n = 4 independent cell lines (B). Representative Western blots of inflammasome markers in VSMC lysates were isolated after 16 h of 20 nM rapamycin treatment (C). Densitometric quantification of protein levels expressed as fold change ± SEM, n = 4 (D). Representative Western blots of inflammasome markers in VSMC lysates after 16 h of 10 nM MitoTEMPO treatment (E). Densitometric quantification of protein levels expressed as fold change ± SEM, n = 4 (F). Estimation of circulating mitochondrial DNA with MTCO1 specific-Taqman probe using qPCR. Data are Ct values (Mean ± SEM, n = 3-4) (G). Representative Western blots of p202 protein levels in VSMC lysates with and without rapamycin or MitoTEMPO treatments (H). Densitometric quantification of protein levels expressed as fold change ± SEM, n = 4. (I). *P < 0.05; **P < 0.01.
The hematopoietic interferon-inducible nuclear protein p202 serves as a dominant-negative antagonist of AIM2, blocking downstream signal transduction by binding to cytoplasmic dsDNA that act as ligands to AIM2[68]. Because none of the antagonists we tested had an effect on AIM2 protein expression, we examined p202 levels in response to antagonist treatment in VSMCs from old LCR rats. Rapamycin increased p202 levels by 3.7-fold [Figure 6H and I]. MitoTEMPO increased p202 levels 2.4-fold, but the effect was not statistically significant. Together, these data support the notion that targeting mtDNA damage and decreasing mtDNA DAMPs prevents inflammasome activation at the intersection of low oxidative capacity and aging.
Rapamycin and MitoTEMPO reduced mitochondrial oxidative stress and increased mitophagy and OCR in VSMCs from old LCR rats
We next examined whether rapamycin or MitoTEMPO had salutary effects on mitophagy. Both rapamycin and MitoTEMPO increased mitophagy by 4.8 and 2.9-fold over respective controls, as measured by colocalization of LAMP1 with TOM20 [Figure 7A and B; Supplementary Figure 8A]. The mitochondrial ROS levels, as assessed by changes in MitoSOX Red fluorescence, decreased by 2.5 and 3-fold in old LCR rat VSMCs treated with rapamycin and MitoTEMPO compared to respective controls [Figure 7C and D, Supplementary Figure 8B], suggesting impaired mitophagy promotes ROS generation[69]. In order to determine whether enhanced mitophagy and reduced mitochondrial ROS levels affected mitochondrial function, we measured OCR in VSMCs from old LCR rats treated with and without rapamycin and MitoTEMPO. Both the compounds significantly increased maximal OCR and reserve respiratory capacity in old LCR rat VSMCs [Figure 7E-H], suggesting the importance of mitochondrial function in vascular health at the intersection of aging and low oxidative capacity.
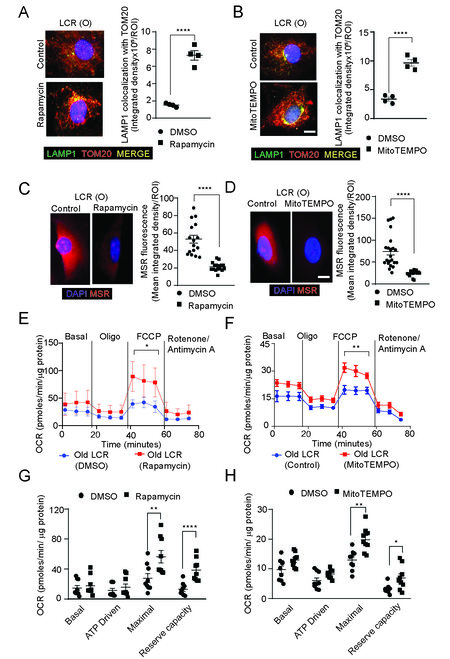
Figure 7. Treatment with rapamycin or MitoTEMPO decreases oxidative stress and enhances mitophagy and OCR in old age LCR rat VSMCs. Representative confocal images of VSMCs treated with and without 20 nM rapamycin (A), or MitoTEMPO (B) and co-stained for lysosomal marker LAMP1 (green) and mitochondrial marker TOM20 (red). Bright yellow fluorescence indicates colocalization of LAMP1 and TOM20 and active mitophagy. Data are presented as average integrated density/ROI ± SEM where n = 4. Scale is 10 μm. Representative confocal images of mitochondrial ROS levels with and without rapamycin (C) or MitoTEMPO (D) treatments as measured by MitoSOX Red (MSR) fluorescence. Intensity of fluorescence was presented as integrated density (mean ± SEM, n = 4, Scale is 10 μm). OCR was measured at the basal level and in the presence of 2 μM oligomycin, 500 nM trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP), and 0.5 μM antimycin A + 500 nM rotenone in VMSCs of old age LCR rats treated without and with rapamycin (E and G) or MitoTEMPO (F and H). Mitochondrial bioenergetic parameters were derived from VSMC OCR measurements.
DISCUSSION
In the present study, we provide experimental evidence that mtDNA damage and dysfunction are induced by low intrinsic aerobic capacity and aging, adversely affecting vascular health. Our results show: (1) increased transcription of genes involved in cell cycle regulation and pro-inflammatory pathways, impaired mitophagy, and impaired oxidative phosphorylation pathways in old age LCR rat aortas versus longevity regulating pathways, increased oxidative phosphorylation and mitophagy/autophagy pathways in old age HCR rat aortas; (2) impaired arterial relaxation and increased aortic stiffness in old age LCR rats compared to the HCR rats; (3) increased mitochondrial oxidative stress and DNA damage in aortas and mitochondrial oxidative stress in aortic VSMCs of old age LCR versus HCR rats; (4) increased expression of AIM2 and its downstream effectors and VCAM1 in the aortic medial layer, as well as AIM2 activation in the aortic VSMCs of old age LCR versus HCR rats; (5) mtDNA DAMP-treated VSMCs from young LCR rats show similar expression and activation of AIM2 inflammasome as do those from old LCR rats; and (6) rapamycin and MitoTEMPO attenuate mtDNA DAMPs-induced inflammasome activation in young LCR rat aortic VSMCs and reduce mitochondrial oxidative stress, improving OCR and mitophagy in old age LCR rat VSMCs. Together, these results demonstrate that mtDNA DAMPs associated with low aerobic capacity contribute to sterile inflammation in aging, resulting in vascular dysfunction and aortic stiffness.
We and others have demonstrated that increased mitochondrial oxidative stress in aging causes arterial stiffening[16,30,40,55]. Seals and colleagues reported that mitochondrial quality control, a balance between mitochondrial biogenesis and mitophagy, is impaired in aging, resulting in higher collagen-1 levels and aortic stiffness[30]. The present data demonstrate that mitochondrial oxidative stress causes VSMC and arterial dysfunction, as evidenced by impaired relaxation of the mesenteric arteries to acetylcholine and aortic stiffening in older LCR rats, extracellular matrix remodeling and fibrosis, and by a leftward shift in the stress-strain curves. The increased production of mitochondrial ROS and inflammation, as evident by increased IL-18 and VCAM1 levels in VSMCs old LCR rats, causes oxidative damage and degradation of elastic laminae, as well as excessive collagen deposition in the vascular wall. Increased vascular stiffness and dysfunction are a direct consequence of these processes[40]. Endothelial dysfunction resulting from increased mitochondrial oxidative stress may also contribute to aortic stiffening in old age LCR rats, as illustrated by decreased eNOS levels, decreased phosphorylation of eNOS (Ser1177), and
Reduced cardiorespiratory fitness is associated with arterial stiffness in coronary artery disease patients[33]. Chronic exercise training in aged rats reduces mitochondrial ROS production, preserving aortic mitochondrial function, and reducing aortic stiffening[75]. Old age HCR rats that demonstrate better mitochondrial energetics, lower ROS levels and less DNA damage in VSMCs than do age-matched LCR rats are protected against aortic stiffening and arterial dysfunction. These results are in general agreement with a study in which cardiomyocytes from HCR rats showed higher aerobic capacity, mitochondrial fitness, and health in association with a longer lifespan[76].
Moreover, our data indicating impaired autophagy and lysosomal dysfunction in aortic VSMCs in old age LCR rats agree with the reports of age-associated decline in proteolytic systems involved in protein quality control, including the autophagy-lysosomal system[77,78]. Aged LCR rats have a significant increase in mitochondrial ROS levels, mtDNA damage and nuclear DNA damage compared with young LCR rats. It is suggested that impaired mitophagy may be caused by reduced ATP levels that inhibit lysosomal function and protein sequestration[79]. When mitochondrial depolarization occurs, PINK1 recruits Parkin to mitochondria and initiates mitophagy. Mitophagy could be compromised by low levels of ATP, which inhibits the upregulation of PINK1 levels and the recruitment of Parkin into mitochondria[80]. As mitophagy genes are encoded by nuclear DNA, impaired mitophagy may reflect nuclear DNA damage in aged LCR rats[81].
Perhaps lower mitochondrial ROS levels and less nuclear and mtDNA damage can explain why mitophagy is not impaired in aged HCR rats. In fact, our RNA-Seq data show upregulation of mitophagy in old HCR rats. In addition, insulin signaling[82] and AMPK signaling[83,84] pathways that are implicated in mitophagy activation are upregulated in old HCR rats. Further supporting impaired mitophagy in old LCR rats, mitochondrial quality control and biogenesis effectors SIRT1/TFAM are downregulated in aged LCR rats[85], whereas SIRT3 levels in SIRT3/FOXO3A-PINK1-PARKIN axis[86] linked to higher mitophagy are high in HCR rats[87].
Additionally, our findings are also supported by the observation that autophagy/mitophagy and mitochondrial fitness are better preserved in HCR rats compared to LCR rats[76]. An impaired mitophagy, especially in a context of pronounced energy deficits, such as in the LCR rat aortas, causes an accumulation of fragments of the mitochondrial genome, such as DAMPs, resulting in sterile inflammation[88]. In fact, higher DAMP levels in aging have been proposed as a potential mechanism for inflammaging[89]. As per conventional wisdom, various pathways of mitochondrial dysfunction converge to activate NLRP3 inflammasomes, but not other types of inflammasomes[90,91]. In contrast, our results indicate higher AIM2 protein levels and AIM2 inflammasome activation in aortic medial VSMCs and isolated aortic VSMCs of old age LCR rats. Interestingly, AIM2 protein expression as well as AIM2 inflammasome activity were both increased in young LCR aortic VSMCs treated with mtDNA DAMPs in culture, supporting the notion that mtDNA DAMPs are inflammaging inducers.
Both rapamycin and MitoTEMPO inhibited AIM2 inflammasome activation but did not inhibit its expression. Rapamycin can reverse age-related vascular dysfunction and increase longevity in rodents[67]. The inhibitory effect of rapamycin[64] on AIM2 activation was attributed to their autophagy/mitophagy-promoting actions. Interestingly, MitoTEMPO was able to inhibit mtDNA DAMP-induced AIM2 inflammasome activity and rescue mitophagy, perhaps by reducing mtROS levels. Additionally, both treatments enhanced the mitochondrial respiratory capacity of VSMCs from old-age LCR rats, which might contribute to better vascular function. Furthermore, the treatments inhibited mtDNA damage in aortic VSMCs in aging, possibly disrupting the nexus between DNA damage and inflammasome activation[92]. Rapamycin increased p202 levels, which may block DAMPs binding to AIM2 inflammasomes[68]. These findings are supported by the observation that rapamycin[66] and MitoTEMPO[93] inhibit the activity of AIM2 inflammasomes. We and others have shown that rapamycin and MitoTEMPO are beneficial against aortic stiffening in mouse models of oxidative stress[40,67].
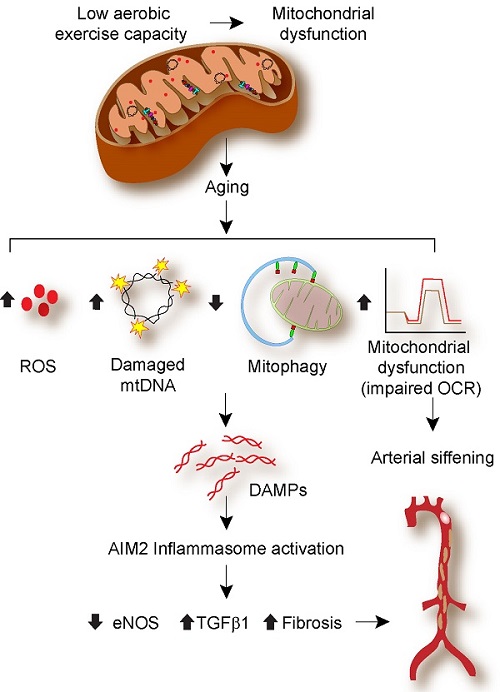
In summary, our data demonstrate that low aerobic capacity and aging cause mitochondrial dysfunction and increase mtROS levels, impair autophagy and mitophagy, resulting in increased inflammasome activation. Mitochondrial turnover in aging is intricately intertwined with inflammasome activation, affecting mitochondrial fitness, VSMC and endothelial function, and vascular health [Figure 8]. Mitophagy induction and inflammasome inhibition, as well as the use of mitochondrial antioxidants, may prove to be helpful therapeutic strategies for enhancing vascular health in older individuals with a reduced aerobic capacity.
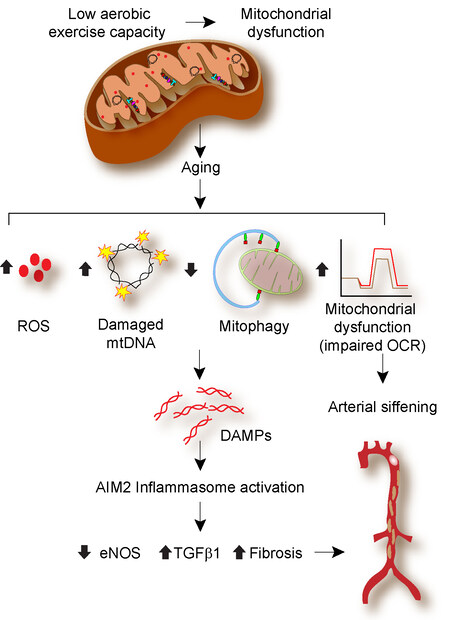
Figure 8. Schematic representation of the low aerobic capacity and aging leading to arterial stiffening.
DECLARATIONS
Authors’ contributionsDesigned the study and experiments: Canugovi C, Madamanchi NR
Performed the experiments: Canugovi C, Stevenson MD, Vendrov AE, Lozhkin A
Wrote the manuscript: Canugovi C, Britton SL, Koch LG, Runge MS, Madamanchi NR
Availability of data and materialsData will be provided on request.
Financial support and sponsorshipNone.
Conflicts of interestDr. Marschall Runge is a member of the Board of Directors at Eli Lilly and Company. Other authors declared that there are no conflicts of interest.
Ethical approval and consent to participateAll animal procedures were performed in compliance with the protocols approved by the Institutional Animal Care and Use Committee of the University of Michigan (PRO00010178) in accordance with NIH OLAW guidelines.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
Supplementary MaterialsREFERENCES
1. Blair SN, Kampert JB, Kohl HW, et al. Influences of cardiorespiratory fitness and other precursors on cardiovascular disease and all-cause mortality in men and women. JAMA 1996;276:205-10.
2. Church TS, Cheng YJ, Earnest CP, et al. Exercise capacity and body composition as predictors of mortality among men with diabetes. Diabetes Care 2004;27:83-8.
3. Gulati M, Pandey DK, Arnsdorf MF, et al. Exercise capacity and the risk of death in women: the St James Women Take Heart Project. Circulation 2003;108:1554-9.
4. Ladenvall P, Persson CU, Mandalenakis Z, et al. Low aerobic capacity in middle-aged men associated with increased mortality rates during 45 years of follow-up. Eur J Prev Cardiol 2016;23:1557-64.
5. Myers J, Prakash M, Froelicher V, Do D, Partington S, Atwood JE. Exercise capacity and mortality among men referred for exercise testing. N Engl J Med 2002;346:793-801.
6. Fleg JL, Strait J. Age-associated changes in cardiovascular structure and function: a fertile milieu for future disease. Heart Fail Rev 2012;17:545-54.
7. Britton SL, Koch LG. Animal genetic models for complex traits of physical capacity. Exerc Sport Sci Rev 2001;29:7-14.
8. Koch LG, Britton SL. Artificial selection for intrinsic aerobic endurance running capacity in rats. Physiol Genomics 2001;5:45-52.
9. Koch LG, Kemi OJ, Qi N, et al. Intrinsic aerobic capacity sets a divide for aging and longevity. Circ Res 2011;109:1162-72.
10. Wisløff U, Najjar SM, Ellingsen O, et al. Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science 2005;307:418-20.
11. Esposito LA, Melov S, Panov A, Cottrell BA, Wallace DC. Mitochondrial disease in mouse results in increased oxidative stress. Proc Natl Acad Sci USA 1999;96:4820-5.
12. Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res 2007;100:460-73.
13. Bonnard C, Durand A, Peyrol S, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest 2008;118:789-800.
14. Yokota T, Kinugawa S, Hirabayashi K, et al. Oxidative stress in skeletal muscle impairs mitochondrial respiration and limits exercise capacity in type 2 diabetic mice. Am J Physiol Heart Circ Physiol 2009;297:H1069-77.
15. Mercer JR, Cheng KK, Figg N, et al. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ Res 2010;107:1021-31.
16. Canugovi C, Stevenson MD, Vendrov AE, et al. Increased mitochondrial NADPH oxidase 4 (NOX4) expression in aging is a causative factor in aortic stiffening. Redox Biol 2019;26:101288.
17. Madamanchi NR, Hakim ZS, Runge MS. Oxidative stress in atherogenesis and arterial thrombosis: the disconnect between cellular studies and clinical outcomes. J Thromb Haemost 2005;3:254-67.
18. Maynard S, Fang EF, Scheibye-Knudsen M, Croteau DL, Bohr VA. DNA damage, DNA repair, aging, and neurodegeneration. Cold Spring Harb Perspect Med 2015;5:a025130.
19. Moon SK, Thompson LJ, Madamanchi N, et al. Aging, oxidative responses, and proliferative capacity in cultured mouse aortic smooth muscle cells. Am J Physiol Heart Circ Physiol 2001;280:H2779-88.
20. Vendrov AE, Stevenson MD, Alahari S, et al. Attenuated superoxide dismutase 2 activity induces atherosclerotic plaque instability during aging in hyperlipidemic mice. J Am Heart Assoc 2017;6:e006775.
21. Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 2005;8:3-5.
22. Kubli DA, Gustafsson ÅB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res 2012;111:1208-21.
23. Caielli S, Athale S, Domic B, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med 2016;213:697-713.
24. Choudhuri S, Chowdhury IH, Garg NJ. Mitochondrial regulation of macrophage response against pathogens. Front Immunol 2020;11:622602.
25. Picca A, Lezza AMS, Leeuwenburgh C, et al. Circulating mitochondrial DNA at the crossroads of mitochondrial dysfunction and inflammation during aging and muscle wasting disorders. Rejuvenation Res 2018;21:350-9.
26. Picca A, Lezza AMS, Leeuwenburgh C, et al. Fueling inflamm-aging through mitochondrial dysfunction: mechanisms and molecular targets. Int J Mol Sci 2017;18:933.
27. Kapetanovic R, Bokil NJ, Sweet MJ. Innate immune perturbations, accumulating DAMPs and inflammasome dysregulation: a ticking time bomb in ageing. Ageing Res Rev 2015;24:40-53.
28. Jheng HF, Tsai PJ, Guo SM, et al. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol 2012;32:309-19.
29. Marseglia L, Manti S, D’Angelo G, et al. Oxidative stress in obesity: a critical component in human diseases. Int J Mol Sci 2014;16:378-400.
30. LaRocca TJ, Hearon CM Jr, Henson GD, Seals DR. Mitochondrial quality control and age-associated arterial stiffening. Exp Gerontol 2014;58:78-82.
31. Zanoli L, Briet M, Empana JP, et al. Vascular consequences of inflammation: a position statement from the ESH Working Group on Vascular Structure and Function and the ARTERY Society. J Hypertens 2020;38:1682-98.
32. Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part I: aging arteries: a “set up” for vascular disease. Circulation 2003;107:139-46.
33. Alves AJ, Oliveira NL, Lopes S, et al. Arterial stiffness is related to impaired exercise capacity in patients with coronary artery disease and history of myocardial infarction. Heart Lung Circ 2019;28:1614-21.
34. Vaitkevicius PV, Fleg JL, Engel JH, et al. Effects of age and aerobic capacity on arterial stiffness in healthy adults. Circulation 1993;88:1456-62.
35. Aroor AR, Jia G, Sowers JR. Cellular mechanisms underlying obesity-induced arterial stiffness. Am J Physiol Regul Integr Comp Physiol 2018;314:R387-98.
36. Bender SB, Castorena-Gonzalez JA, Garro M, et al. Regional variation in arterial stiffening and dysfunction in Western diet-induced obesity. Am J Physiol Heart Circ Physiol 2015;309:H574-82.
37. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25:1105-11.
38. Ge SX, Son EW, Yao R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform 2018;19:534.
39. Raudvere U, Kolberg L, Kuzmin I, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 2019;47:W191-8.
40. Vendrov AE, Vendrov KC, Smith A, et al. NOX4 NADPH oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid Redox Signal 2015;23:1389-409.
41. Akula S, Brosch IK, Leipzig ND. Fluorinated methacrylamide chitosan hydrogels enhance cellular wound healing processes. Ann Biomed Eng 2017;45:2693-702.
42. Santos JH, Meyer JN, Mandavilli BS, Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. In: Henderson DS, editor. DNA Repair Protocols. Humana Press: Totowa. 2006. pp. 183-99.
43. Arribas SM, Briones AM, Bellingham C, et al. Heightened aberrant deposition of hard-wearing elastin in conduit arteries of prehypertensive SHR is associated with increased stiffness and inward remodeling. Am J Physiol Heart Circ Physiol 2008;295:H2299-307.
44. Gutiérrez-Arzapalo PY, Rodríguez-Rodríguez P, Ramiro-Cortijo D, et al. Role of fetal nutrient restriction and postnatal catch-up growth on structural and mechanical alterations of rat aorta. J Physiol 2018;596:5791-806.
45. Laurent S, Boutouyrie P, Asmar R, et al. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension 2001;37:1236-41.
46. Sutton-Tyrrell K, Najjar SS, Boudreau RM, et al. Elevated aortic pulse wave velocity, a marker of arterial stiffness, predicts cardiovascular events in well-functioning older adults. Circulation 2005;111:3384-90.
47. Mitchell GF, Hwang SJ, Vasan RS, et al. Arterial stiffness and cardiovascular events: the framingham heart study. Circulation 2010;121:505-11.
48. Soucy KG, Ryoo S, Benjo A, et al. Impaired shear stress-induced nitric oxide production through decreased NOS phosphorylation contributes to age-related vascular stiffness. J Appl Physiol 2006;101:1751-9.
49. Fleenor BS, Marshall KD, Durrant JR, Lesniewski LA, Seals DR. Arterial stiffening with ageing is associated with transforming growth factor-β1-related changes in adventitial collagen: reversal by aerobic exercise. J Physiol 2010;588:3971-82.
50. Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol 2005;25:932-43.
51. Saura M, Zaragoza C, Herranz B, et al. Nitric oxide regulates transforming growth factor-beta signaling in endothelial cells. Circ Res 2005;97:1115-23.
52. Crosas-Molist E, Meirelles T, López-Luque J, et al. Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arterioscler Thromb Vasc Biol 2015;35:960-72.
53. Gioscia-Ryan RA, Battson ML, Cuevas LM, Eng JS, Murphy MP, Seals DR. Mitochondria-targeted antioxidant therapy with MitoQ ameliorates aortic stiffening in old mice. J Appl Physiol 2018;124:1194-202.
54. Rossman MJ, Santos-Parker JR, Steward CAC, et al. Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension 2018;71:1056-63.
55. Zhou RH, Vendrov AE, Tchivilev I, et al. Mitochondrial oxidative stress in aortic stiffening with age: the role of smooth muscle cell function. Arterioscler Thromb Vasc Biol 2012;32:745-55.
56. Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res 2012;95:194-204.
57. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 2004;84:767-801.
58. Ballinger SW, Patterson C, Knight-Lozano CA, et al. Mitochondrial integrity and function in atherogenesis. Circulation 2002;106:544-9.
59. Wenzel P, Schuhmacher S, Kienhöfer J, et al. Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc Res 2008;80:280-9.
60. Crowley LC, Christensen ME, Waterhouse NJ. Measuring mitochondrial transmembrane potential by TMRE staining. Cold Spring Harb Protoc 2016;2016:pdb.prot087361.
61. Stuart JA, Brown MF. Mitochondrial DNA maintenance and bioenergetics. Biochim Biophys Acta 2006;1757:79-89.
62. Iwashita H, Torii S, Nagahora N, et al. Live cell imaging of mitochondrial autophagy with a novel fluorescent small molecule. ACS Chem Biol 2017;12:2546-51.
63. Dai J, Zhang X, Li L, Chen H, Chai Y. Autophagy inhibition contributes to ROS-producing NLRP3-dependent inflammasome activation and cytokine secretion in high glucose-induced macrophages. Cell Physiol Biochem 2017;43:247-56.
64. Ponomareva L, Liu H, Duan X, et al. AIM2, an IFN-inducible cytosolic DNA sensor, in the development of benign prostate hyperplasia and prostate cancer. Mol Cancer Res 2013;11:1193-202.
65. Kong DH, Kim YK, Kim MR, Jang JH, Lee S. Emerging roles of vascular cell adhesion molecule-1 (VCAM-1) in immunological disorders and cancer. Int J Mol Sci 2018;19:1057.
66. Wang Y, Li Z, Teng M, Liu J. Dihydroartemisinin inhibits activation of the AIM2 inflammasome pathway and NF-κB/HIF-1α/VEGF pathway by inducing autophagy in A431 human cutaneous squamous cell carcinoma cells. Int J Med Sci 2021;18:2705-15.
67. Lesniewski LA, Seals DR, Walker AE, et al. Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell 2017;16:17-26.
68. Ru H, Ni X, Zhao L, et al. Structural basis for termination of AIM2-mediated signaling by p202. Cell Res 2013;23:855-8.
69. Linton PJ, Gurney M, Sengstock D, Mentzer RM Jr, Gottlieb RA. This old heart: cardiac aging and autophagy. J Mol Cell Cardiol 2015;83:44-54.
70. Kahveci AS, Barnatan TT, Kahveci A, et al. Oxidative stress and mitochondrial abnormalities contribute to decreased endothelial nitric oxide synthase expression and renal disease progression in early experimental polycystic kidney disease. Int J Mol Sci 2020;21:1994.
71. Morris EM, Meers GME, Ruegsegger GN, et al. Intrinsic high aerobic capacity in male rats protects against diet-induced insulin resistance. Endocrinology 2019;160:1179-92.
72. Sansbury BE, Cummins TD, Tang Y, et al. Overexpression of endothelial nitric oxide synthase prevents diet-induced obesity and regulates adipocyte phenotype. Circ Res 2012;111:1176-89.
73. Valerio A, Cardile A, Cozzi V, et al. TNF-α downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. J Clin Invest 2006;116:2791-8.
74. Su X, Feng X, Terrando N, et al. Dysfunction of inflammation-resolving pathways is associated with exaggerated postoperative cognitive decline in a rat model of the metabolic syndrome. Mol Med 2013;18:1481-90.
75. Gu Q, Wang B, Zhang XF, Ma YP, Liu JD, Wang XZ. Chronic aerobic exercise training attenuates aortic stiffening and endothelial dysfunction through preserving aortic mitochondrial function in aged rats. Exp Gerontol 2014;56:37-44.
76. Aon MA, Cortassa S, Juhaszova M, et al. Mitochondrial health is enhanced in rats with higher vs. lower intrinsic exercise capacity and extended lifespan. NPJ Aging Mech Dis 2021;7:1.
77. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013;153:1194-217.
78. Tomaru U, Takahashi S, Ishizu A, et al. Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am J Pathol 2012;180:963-72.
80. Lee S, Zhang C, Liu X. Role of glucose metabolism and ATP in maintaining PINK1 levels during Parkin-mediated mitochondrial damage responses. J Biol Chem 2015;290:904-17.
81. D’Amico D, Mottis A, Potenza F, et al. The RNA-binding protein PUM2 impairs mitochondrial dynamics and mitophagy during aging. Mol Cell 2019;73:775-787.e10.
82. Galizzi G, Palumbo L, Amato A, et al. Altered insulin pathway compromises mitochondrial function and quality control both in in vitro and in vivo model systems. Mitochondrion 2021;60:178-88.
83. Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011;331:456-61.
84. Wang XD, Yu WL, Sun Y. Activation of AMPK restored impaired autophagy and inhibited inflammation reaction by up-regulating SIRT1 in acute pancreatitis. Life Sci 2021;277:119435.
85. Choi J, Chandrasekaran K, Demarest TG, et al. Brain diabetic neurodegeneration segregates with low intrinsic aerobic capacity. Ann Clin Transl Neurol 2014;1:589-604.
86. Wan W, Hua F, Fang P, et al. Regulation of mitophagy by sirtuin family proteins: a vital role in aging and age-related diseases. Front Aging Neurosci 2022;14:845330.
87. Karvinen S, Silvennoinen M, Vainio P, et al. Effects of intrinsic aerobic capacity, aging and voluntary running on skeletal muscle sirtuins and heat shock proteins. Exp Gerontol 2016;79:46-54.
88. Kauppila TES, Kauppila JHK, Larsson NG. mammalian mitochondria and aging: an update. Cell Metab 2017;25:57-71.
89. Goldberg EL, Dixit VD. Drivers of age-related inflammation and strategies for healthspan extension. Immunol Rev 2015;265:63-74.
90. Cruz CS, Kang MJ. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 2018;41:37-44.
91. Gurung P, Lukens JR, Kanneganti TD. Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends Mol Med 2015;21:193-201.
92. Licandro G, Ling Khor H, Beretta O, et al. The NLRP3 inflammasome affects DNA damage responses after oxidative and genotoxic stress in dendritic cells. Eur J Immunol 2013;43:2126-37.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Canugovi C, Stevenson MD, Vendrov AE, Lozhkin A, Britton SL, Koch LG, Runge MS, Madamanchi NR. Mitochondrial DAMPs-dependent inflammasome activation during aging induces vascular smooth muscle cell dysfunction and aortic stiffness in low aerobic capacity rats. J Cardiovasc Aging 2022;2:47. http://dx.doi.org/10.20517/jca.2022.35
AMA Style
Canugovi C, Stevenson MD, Vendrov AE, Lozhkin A, Britton SL, Koch LG, Runge MS, Madamanchi NR. Mitochondrial DAMPs-dependent inflammasome activation during aging induces vascular smooth muscle cell dysfunction and aortic stiffness in low aerobic capacity rats. The Journal of Cardiovascular Aging. 2022; 2(4): 47. http://dx.doi.org/10.20517/jca.2022.35
Chicago/Turabian Style
Canugovi, Chandrika, Mark D. Stevenson, Aleksandr E. Vendrov, Andrey Lozhkin, Steven L. Britton, Lauren G. Koch, Marschall S. Runge, Nageswara R. Madamanchi. 2022. "Mitochondrial DAMPs-dependent inflammasome activation during aging induces vascular smooth muscle cell dysfunction and aortic stiffness in low aerobic capacity rats" The Journal of Cardiovascular Aging. 2, no.4: 47. http://dx.doi.org/10.20517/jca.2022.35
ACS Style
Canugovi, C.; Stevenson MD.; Vendrov AE.; Lozhkin A.; Britton SL.; Koch LG.; Runge MS.; Madamanchi NR. Mitochondrial DAMPs-dependent inflammasome activation during aging induces vascular smooth muscle cell dysfunction and aortic stiffness in low aerobic capacity rats. J. Cardiovasc. Aging. 2022, 2, 47. http://dx.doi.org/10.20517/jca.2022.35
About This Article
Copyright

Data & Comments
Data



 Cite This Article 32 clicks
Cite This Article 32 clicks

 Like This Article 15
likes
Like This Article 15
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.