
Metabolic targets in cardiac aging and rejuvenation

 , ...
, ... Abstract
Cardiac aging is accompanied by progressive loss of cellular function, leading to impaired heart function and heart failure. There is an urgent need for efficient strategies to combat this age-related cardiac dysfunction. A growing number of events suggest that age-related cardiac diseases are tightly related to metabolic imbalance. This review summarizes recent findings concerning metabolic changes during cardiac aging and highlights the therapeutic approaches that target metabolic pathways in cardiac aging.
Keywords
INTRODUCTION
Due to medical progress and lifestyle changes, life expectancy has been significantly prolonged worldwide. However, multiple diseases, such as cardiovascular diseases (CVD) and metabolic disorders, tend to occur at older ages. For example, the incidence of CVDs, including hypertension, coronary heart disease (CHD), and heart failure (HF), is two-fold higher in the population over 80 years old compared with those who are 40 years of age[1]. The incidence of myocardial infarction (MI) increases sevenfold in the elderly population aged 70 years compared to those aged 40 years[2]. Aging is well recognized as one of the critical risk factors for heart disease. Nearly two-thirds of those suffering from cardiovascular disease are elderly patients[3]. As the aging population worldwide is growing at a remarkable rate, anti-aging strategies to improve cardiovascular health and lifespan are imminently required. Therefore, understanding the mechanisms of cardiac aging is vital for the therapeutic development of CVDs in the elderly population.
In the aging process, the heart function degenerates gradually and may eventually lead to HF. Significant structural alteration of the left ventricle and an increase in fibrosis is observed in aged hearts. Moreover, diastolic dysfunction and systolic dysfunction are prevalent in aged hearts. Another consequence is the decline of the cardiac reserves in aged hearts, which contributes to HF with preserved ejection fraction (HFpEF), the most common type of HF in the aged population[4,5]. Although the cardiac physical changes of aging are well characterized, the intrinsic features and pathways driving the age-associated decline of heart function are not fully understood. Intrinsic features, such as mitochondrial dysfunction, inflammation, and reactive oxygen species (ROS), were considered significant drivers of cardiac aging [Figure 1]. In the aging process, there is a metabolic decline and disruption of nutrient uptake by body tissues. Almost all the hallmarks of aging are affected by cellular metabolic disorders. Mitochondrial metabolism and metabolic pathways were proved to play important roles in cardiac aging. The aged heart exhibits impaired metabolic flexibility, reduced ability to oxidize fatty acids, and an enhanced dependence on glucose metabolism[6]. Therefore, deciphering the molecular mechanisms underlying cardiac metabolic dysfunction could reveal potential interventional targets to attenuate cardiac degeneration caused by aging.

Figure 1. Metabolic pathways and regulators affect the hallmarks of aging in the heart. Cx43: Connexin-43; ROS: reactive oxygen species; SASP: senescence-associated secretory phenotype; EPCs: endothelial progenitor cells; AMPK: AMP-activated protein kinase.
This review focuses on the current understanding of metabolic changes and their effects on myocardial aging and discusses the metabolic signaling pathways and metabolites involved in the myocardial aging process, which may provide a roadmap for cardiac rejuvenation and novel therapies for preventing aging-related heart diseases.
METABOLIC CHANGES IN CARDIAC AGING
During aging, cellular homeostasis and body function are progressively dysregulated, which could be determined by several cellular and molecular hallmarks of aging[7]. These hallmarks include genomic instability, telomere attrition, epigenetic alteration, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication[7]. Metabolic pathways and regulators were proved to affect all the hallmarks of aging in the heart [Figure 1].
Mitochondria dysfunction in cardiac aging
Mitochondria play a central role in utilizing nutrient materials and energy production. The energy required by the heart is primarily derived from fatty acid oxidation and subsequent ATP production within the mitochondria. The myocardium is highly susceptible to mitochondrial dysfunction due to heavy dependence on mitochondrial oxidative metabolism[8]. Mitochondrial dysfunction can be induced by several pathways related to senescence, such as DNA damage and telomere attrition[9,10]. Mitochondria in aged cardiomyocytes often present an abnormal structure and an increased ROS level. This increased oxidative stress causes a gradual accumulation of mitochondrial damage and electron transport chain (ETC) dysfunction. These changes are associated with diastolic cardiac dysfunction and left ventricular hypertrophy[11]. Therefore, mitochondrial function changes are considered major contributing factors to cardiac senescence[7].
Mitochondria are the major source and target of ROS in cardiomyocytes[12]. Enlarged mitochondria and higher ROS production have been observed in cardiac aging[13]. In particular, the ETC contributes to ROS generation. Oxidative stress induced by high levels of ROS could cause DNA damage and induce mitochondria in the aged heart to undergo permeability transition, accelerate cytochrome C release, and subsequently initiate programmed cell death[14] [Figure 2]. Moreover, the increased oxidative stress on mitochondria may disrupt cellular homeostasis and create a proinflammatory environment that accelerates aging in mice[15]. Mechanically, the damaged cardiomyocytes exhibit the senescence-associated secreting phenotype (SASP). SASP factors released by senescent cardiomyocytes include CCN family member 1 (CCN1), interleukins (IL1α, IL1β, and IL6), tumor necrosis factor-alpha (TNFα), etc.[16]. Enhanced SASP further promotes the formation of a proinflammatory microenvironment and cardiac aging [Figure 1]. Moreover, increased ROS could trigger reduced expression and pathological redistribution of connexin-43 (Cx43)[17,18] [Figure 1]. Cx43 is the main component of the gap junction in the heart and mediates cellular communication and conduction. The altered distribution of Cx43 may lead to lethal cardiac arrhythmias[19]. The increased ROS level could also cause telomere attrition and inhibit telomerase activity [Figure 1]. Interestingly, the administration of the powerful antioxidant epigallocatechin gallate (EGCG) could reverse the decreased telomere length observed in heart/muscle-specific manganese superoxide dismutase-deficient mice[20].
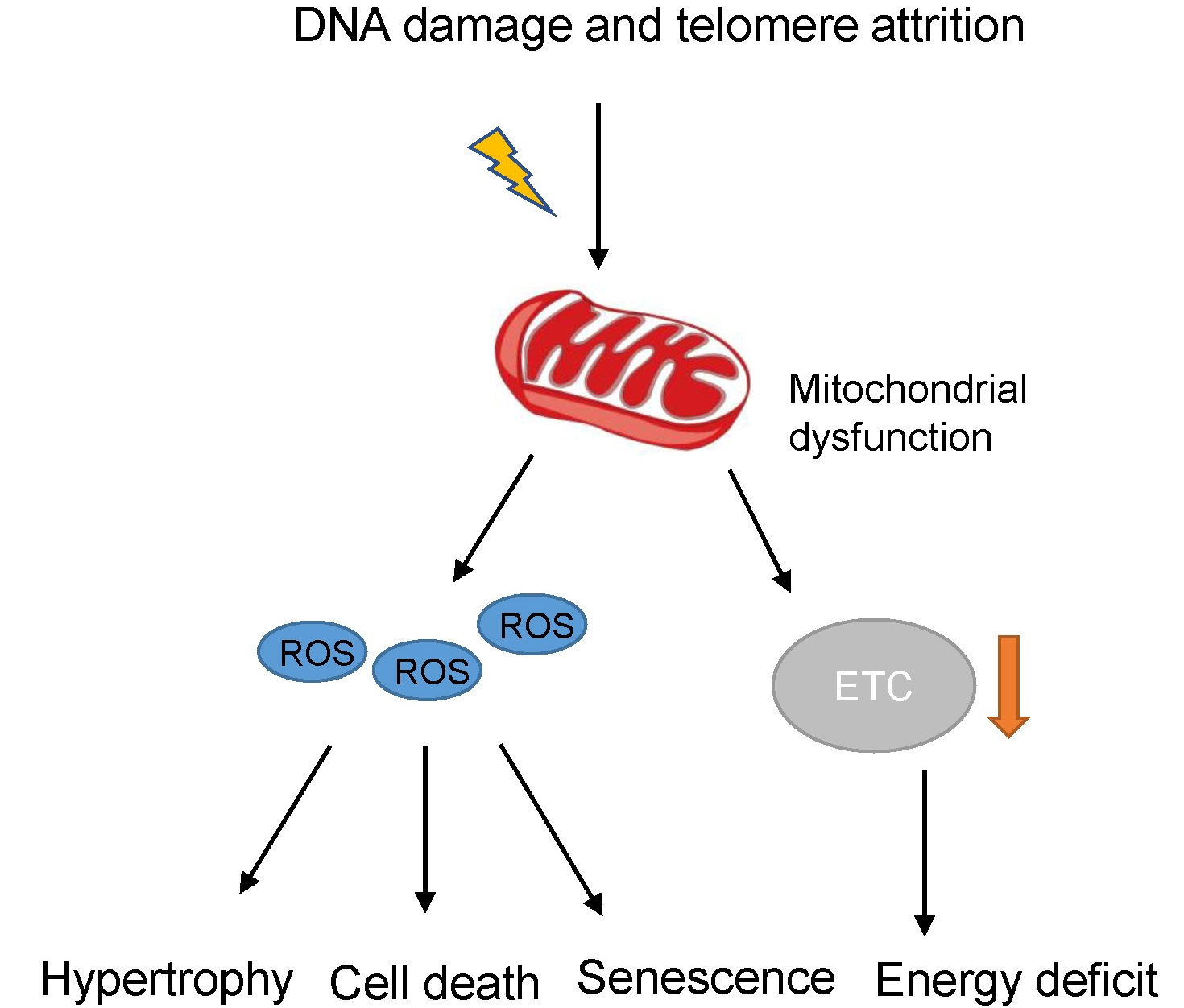
Figure 2. Mitochondria dysfunction in cardiac aging. ROS: Reactive oxygen species; ETC: electron transport chain.
Aging also impairs mitochondrial oxidative phosphorylation (OXPHOS), the main energy source for cardiac tissue. With age, there is a decline in the activity of complexes III and IV, contributing to the decrease in respiration[6]. Moreover, the uptake of fatty acids is increased, while fatty acid oxidation (FAO) is decreased in aged hearts. The heart generates approximately 70% of its energy from FAO and 30% from carbohydrate metabolism under physiological conditions. β-oxidation of fatty acids primarily occurs in the matrix of mitochondria. β-oxidation of fatty acids could produce nicotinamide adenine dinucleotide, reduced form (NADH). As a metabolic intermediate, glycolysis produces pyruvate in the cytoplasm and then enters the mitochondria[21]. Pyruvate is subsequently metabolized by pyruvate dehydrogenase (PDH) to generate NADH and acetyl-CoA in the mitochondrial matrix. In the aging heart, there is an increased capacity for glucose oxidation by mitochondria mediated by enhanced PDH complex activity[22]. Therefore, the fuel preference between fatty acids and glucose shifts in the aging heart, increasing glucose oxidation at the expense of FAO. During aging and diseases, the metabolic alteration in myocardium is essentially associated with impaired cardiac function[23,24]. In failing hearts, the myocardial energy substrate switches from fatty acids to glucose for ATP production[25,26]. Together, mitochondrial dysfunction and metabolic remodeling contribute to the senescence of cardiomyocytes and cardiac aging.
NAD+ metabolism in cardiac aging
Nicotinamide adenine dinucleotide, oxidized form (NAD+), is an essential metabolite in cardiac energy and reduction-oxidation (redox) homeostasis. The heart has a high level of NAD+, and precise control of this metabolite is critical for the cardiac bioenergetic process.
In mammals, NAD+ is synthesized by two different routes: the deamidated and amidated routes. The deamidated route uses the amino acid tryptophan (Trp) to synthesize NAD+ de Novo [Figure 3]. In the deamidated route, nicotinic acid mononucleotide (NaMN) is converted to nicotinic acid dinucleotide (NaAD) by nicotinamide mononucleotide adenyltransferase (NMNAT), and NaAD is then converted to NAD+ by NAD synthase (NADS) [Figure 3]. In the amidated route, nicotinamide riboside kinase (NRK) converts nicotinamide riboside (NR) to nicotinamide mononucleotide (NMN), while nicotinamide phosphoribosyltransferase (NAMPT) activity converts nicotinamide (NAM) to NMN [Figure 3]. The heart lacks the enzymes necessary for the de novo biosynthesis of NAD+. Instead, cardiac cell salvage NAD+ from NAM and NR through the amidated route (salvage pathway)[27] [Figure 3]. Nearly all cardiac NAD+ is generated through the salvage pathway[27]. Therefore, the salvage pathway is dominant in the heart for providing the NAD+ required to maintain the high metabolic demands.

Figure 3. Biosynthesis of NAD+. Trp: Tryptophan; NaMN: nicotinic acid mononucleotide; NMNAT: nicotinamide mononucleotide adenyltransferase; NaAD: nicotinic acid dinucleotide; NADS: NAD synthase; NAM: nicotinamide; NR: nicotinamide riboside; NAMPT: nicotinamide phosphoribosyltransferase; NRK: nicotinamide riboside kinase; NMN: nicotinamide mononucleotide.
NAD+ cannot cross the plasma membrane by passive transport because of its size and positive charge. Therefore, cardiac cells import NAD+ precursors for NAD+ synthesis. Among NAD+ precursors, NAM is the smallest and can cross the plasma membrane by passive transport[28] [Figure 4]. CD73-ecto-5′-nucleotidase (CD73) is responsible for the hydrolysis of extracellular NAD+ to NMN and AMP, and then NMN to NR. NR is transported into cells through solute carrier family 29 members 1/2/4 (SLC29A1/2/4)[29]. Recent studies suggest that the cation/chloride cotransporter solute carrier family 12 member 8 (SLC12A8) is a specific NMN transporter[30] [Figure 4]. Moreover, SLC25A51 was recently discovered as a mammalian mitochondrial NAD+ transporter[31]. Further studies are required to clarify the transport mechanisms of NAD+ and its precursors in the cardiovascular system.

Figure 4. Metabolism of NAD+. NAM: Nicotinamide; NMN: nicotinamide mononucleotide; ETC: electron transport chain; TCA: tricarboxylic acid cycle; NAMPT: nicotinamide phosphoribosyltransferase; NR: nicotinamide riboside; PARP: poly(ADP-ribose) polymerase.
Intracellular NAD+ concentrations decline with age in multiple organs, including the heart[32]. The NAD+ concentration decline might be because of a decrease in NAD+ biosynthesis and increased NAD+ degradation. Indeed, age-related downregulation of NAMPT has been observed in mice and humans[33], which may affect systemic NAD+ levels. For NAD+ degradation, growing evidence suggests that CD38-NAD+-glycohydrolase (CD38) contributes to age-related NAD+ decline in mammals[34,35]. CD38 hydrolyzes NAD+ to NAM and ADP-ribose (ADPR) or nicotinamide mononucleotide (NMN) to NAM. Interestingly, a CD38 inhibitor reverses age-related NAD+ degradation and improves cardiac function in aged mice[36]. CD38 also exhibits an activity that degrades circulating NMN in vivo[34]. Therefore, coadministration of NAD+ precursors and CD38 antagonists might be more efficient than NAD+ precursors alone for cardiac anti-aging therapy. CD38 is predominantly expressed in immune cells[37], and the proinflammatory cytokines secreted by senescent cells have been shown to elevate CD38 levels and promote the age-associated decline of NAD+ and NMN[35,38]. In addition to CD38, poly(ADP-ribose) polymerases (PARPs) consume NAD+ to repair age-related DNA damage in aging tissues[39]. Cardiomyocytes are exposed to accumulating metabolic and oxidative damage, which eventually causes DNA damage and PARP activation, thereby reducing NAD+ concentration in the aging heart[40].
NAD+ levels have important biological functions in aging. During aging, the declined cellular NAD+ level can affect DNA repair, epigenetic regulation, autophagy, and redox balance[41] [Figure 1]. Because NAD+ is a cofactor for various enzymes, loss of NAD+ impacts many cellular processes. For example, NAD+ is required for the activity of epigenetic regulators such as histone deacetylase SIRT1, and a decline in its level causes changes in histone acetylation, which subsequently influences chromatin organization and gene expression[41] [Figure 1]. NAD+ is also required for DNA repair via PARPs during aging, and the decline of NAD+ could cause DNA damage accumulation[41]. Autophagy is regulated by NAD+ levels via sirtuins (mostly SIRT1). The decline of NAD+ levels reduces overall autophagy[41] [Figure 1]. Moreover, NAD+ is an important coenzyme in redox reactions. The NAD+/NADH redox balance is required for metabolic homeostasis. Recent evidence suggests that redox-cycling quinone β-lapachone, an exogenous co-substrate of NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), could regenerate NAD+ from NADH[42]. However, β-lapachone administration induces an imbalance of redox cycle and oxidative stresses in some solid tumors and should be administered with caution[43].
Core metabolic regulators in cardiac aging
Sirtuins were initially identified as deacetylases that remove acetyl-lysine modification[44]. However, recent studies suggest that they can also remove acyl-lysine modifications such as malonyl-lysine[45], succinyl-lysine[45], and glutaryl-lysine[46]. In mammals, seven sirtuins (SIRT1-7) have different subcellular locations. SIRT1, SIRT6, and SIRT7 are in the nucleus, while SIRT2 resides primarily in the cytoplasm. SIRT3, SIRT4, and SIRT5 are localized in mitochondria [Figure 4]. SIRT3 is a lysine deacetylase involved in lipid metabolism and oxidative stress[47]. SIRT4 influences amino acid metabolism and the tricarboxylic acid cycle[48]. SIRT5 is a lysine demalonylase, desuccinylase[45], and deglutarylase[46], and it is involved in several metabolic pathways.
NAD+ is a major activator of sirtuins; the dependence of sirtuins on NAD+ links sirtuin function to energy metabolism[49]. The effects of SIRT1 on aging and lifespan have been well recognized[50]. Growing evidence links NAD+ supplementation to SIRT1 activation and shows that SIRT1 protects against cardiac aging[51]. The protective effects of SIRT1 in the heart include inhibition of cardiomyocyte apoptosis, reduced inflammation and oxidative stress, and maintenance of energy metabolism[52]. SIRT1 inhibits nuclear factor-kappaB (NF-κB) signaling by deacetylating the p65 subunit of the NF-κB complex, thus repressing NF-κB-induced inflammatory responses in aging[53]. SIRT1 stimulates oxidative energy production via the activation of AMP-activated protein kinase (AMPK)[54], peroxisome proliferator-activated receptor-alpha (PPARα)[55], and peroxisome proliferator-activated receptor-gamma co-activator-1 alpha (PGC-1α)[56] simultaneously. The inhibition of SIRT1 disrupts oxidative energy metabolism in aging-related diseases. Aging also induces a pathological phenotype in the hearts of SIRT5-knockout mice[57]. The shortening fraction and ejection fraction of aged SIRT5-knockout mice were significantly decreased compared to the levels of similar aged wild-type control mice. Interestingly, succinylation and subsequent inhibition of the mitochondrial trifunctional protein α-subunit contribute to this phenotype in the hearts of aging SIRT5-knockout mice.
The mammalian target of rapamycin (mTOR) is an evolutionarily conserved and atypical serine/threonine kinase. The mTOR signaling pathway plays an important role in regulating cell metabolism. mTOR is a serine/threonine protein kinase, which constitutes the catalytic subunit of two distinct complexes known as mTOR complex 1 (mTORC1) and mTORC2[58]. Since mTORC1 activity is aberrantly elevated in some aged cells, this complex has been the focus of investigation for the last decades[59]. Inhibition of the mTOR pathway by rapamycin treatment, genetic inactivation of mTORC1, or calorie restriction has been shown to extend lifespan[60]. Calorie restriction reduces nutrient intake and pushes mTORC1 towards a catabolic direction[60]. Indeed, calorie restriction failed to confer additional longevity benefits in the context of mTORC1 inhibition, suggesting that calorie restriction counteracts aging through the mTORC1 pathway[61]. Mechanically, mTOR integrates energy and nutrient availability to regulate the synthesis of cellular components[60]. Under amino acid replete conditions, Rag-GTPases serve as nutrient sensing machinery to stimulate mTORC1 kinase activity[62]. In contrast, calorie restriction can deplete cellular stores of ATP and trigger the AMPK complex, which inhibits mTORC1[63] [Figure 1]. mTORC1 also suppresses autophagy to prevent the premature breakdown of newly synthesized cellular components [Figure 1]. This inhibition of autophagy allows damaged proteins to accumulate in the cell and accelerates the aging process [Figure 1]. Indeed, mTOR inhibition by nutrient restriction or rapamycin treatment could restore declined autophagic capacity in aging hearts[64]. However, rapamycin also has the disadvantages of side effects, such as anemia and acute nephrotoxicity[65], and needs to be used cautiously.
The functions of metabolic regulators within non-myocytes in cardiac aging
Myocardial tissues consist of cardiomyocytes and non-myocytes, including endothelial cells, fibroblasts, and immune cells[16]. Metabolic changes under pathological conditions could affect the communications between cardiomyocytes and non-myocytes. For example, metabolic dysfunction in cardiomyocytes could induce the activation of fibroblasts[66]. Moreover, SIRT2 overexpression in cardiomyocytes activated the AMPK pathway and reduced aging-associated fibrosis[66]. Metabolic regulators could modulate the function of fibroblasts in cardiac aging. For example, adiponectin activates AMPK signaling and induces collagen remodeling in cardiac fibroblasts[67]. AMPK activation also increases the content of fibroblasts in the infarcted area[68]. Metabolic factors are also critical for the activation of immune cells. For example, AMPK promotes macrophage fatty acid oxidative metabolism and induces inflammatory macrophage activation in cardiac aging[69]. SIRT1 also regulates the function of macrophages and participates in cardiac aging[70]. Moreover, fatty acid metabolism could modulate T cell activity in cardiac aging[71].
During aging, the metabolism of endothelial cells also changes[72], and endothelial metabolism plays a critical role in cardiac aging. For example, Liver kinase B1 (LKB1) is an important regulator of energy homeostasis by activating the AMPK pathway[73]. Endothelial cell-specific LKB1 deletion causes endothelial dysfunction and induces cardiomyocyte hypertrophy[74]. Endothelial progenitor cells (EPCs) are circulating progenitor cell populations with angiogenic potential at sites of ischemia, hypoxia, or injury[75]. During aging, the function of EPCs declines[76]. Interestingly, the restoration of either intracellular NAD+ levels or SIRT1 expression could improve the function of aged EPCs[76,77] [Figure 1]. Therefore, boosting NAD+ levels in EPCs may serve as a possible way to stimulate angiogenesis in aged hearts.
The impact of sex differences on cardiac metabolism in the context of aging
It is well known that CVD mortality rates are lower in women than men[78]. Although CVD mortality rates increase with age in both genders, female HF patients still have significantly better survival rates than male patients in the aged population[79]. Growing evidence suggests that regulatory pathways in aged female and male hearts are different[80,81]. Estrogen is an obvious regulator of this gender difference. The observation that cardiovascular dysfunction increases when estrogen production ceases further supports this notion[82]. However, the gender difference in cardiac aging is complex and cannot be simply attributed to estrogen alone[83].
Interestingly, the cardioprotective effect of estrogen may be mediated by its regulation of metabolic regulators. For example, the SIRT1 and SIRT3 expression levels are lower in elderly female hearts (50-68 years old) than in young female hearts (17-40 years old)[81]. In contrast, no age-associated changes in SIRT1 and SIRT3 expression are observed in male hearts[81]. In addition, the anti-oxidative enzyme superoxide dismutase 2 (SOD2) in aged female hearts was downregulated, whereas it was upregulated in aged male hearts[81]. Mechanically, estrogen could upregulate SOD2 expression[84], and the age-associated changes in the estrogen level in female blood (down) and male blood (up) may be involved in the gender difference in SOD2 expression change[81]. The cardioprotective effect of estrogen could also function through metabolic pathways such as the AMPK pathway[83]. Estrogen activates AMPK by phosphorylation in myocardium[85], which subsequently promotes glucose transport and free fatty acid metabolism. Therefore, higher levels of circulating estrogens in females may contribute to a stronger ability of AMPK activation compared with their male counterparts[83]. This may provide a more robust protective effect when the energy requirement of female patients with age-associated HF increase.
THERAPEUTIC POTENTIAL AND CHALLENGES
Metabolic protection against cardiac aging
The main aim of cardiac anti-aging therapy is to find an effective medicine to reverse the features of aged hearts. Several molecules that prevent known cardiac aging features via modulating metabolic regulators have been described [Table 1]. For example, Alginate oligosaccharide (AOS) has been shown to be an effective agent in alleviating cardiac aging[86]. This agent could improve mitochondrial biogenesis and maintain mitochondrial integrity. In addition, mitochondrially targeted vitamin E (MitoVitE) and mitochondrially targeted coenzyme Q (MitoQ) can target mitochondrial dysfunction[87,88]. Cellular senescence, another important feature in cardiac aging, can be reversed by SIRT6[89]. This effect was achieved by deacetylation of key metabolic regulators PCSK9 (proprotein convertase subtilisin/kexin type 9), which modulates the plasma LDL cholesterol level. Although resting heart function is not significantly altered, diastolic and systolic dysfunction exists in aged hearts[90]. A recent study showed that mitochondrially targeted peptide SS-31 (elamipretide) treatment can reverse diastolic dysfunction in the rodent model[91]. SS-31 was proved to reduce mitochondrial ROS and protein oxidation in aged hearts via targeting cardiolipin (CL), indicating alleviation of mitochondrial oxidative stress as a potential mechanism[91]. Similarly, overexpression of the antioxidant enzyme catalase can improve the old mice’s heart systolic and diastolic function, and this phenotype is partially mediated by mitochondrial oxidative stress[91,92]. β-hydroxybutyrate (βOHB) treatment could attenuate NLPR3 inflammasome formation and antagonize proinflammatory cytokine-triggered mitochondrial dysfunction in aged mice[93]. This protective effect of βOHB is achieved via activation of CS (citrate synthase) and inhibiting fatty acid uptake. Acetylcarnitine treatment could mitigate age-induced metabolic imbalance via improving cardiac OXPHOS levels[94] [Table 1].
Cardiac aging features and metabolic protection
| Aging features | Protective molecule | Protective mechanism | Reference |
| Mitochondrial dysfunction | Alginate oligosaccharide | Improving mitochondrial biogenesis and maintaining the mitochondrial integrity | [86] |
| Cellular senescence | SIRT 6 | Deacylation of key metabolic regulators such as PCSK9 which modulates plasma LDL-cholesterol level | [52] |
| Diastolic dysfunction and systolic dysfunction | Mitochondrial targeted peptide SS-31 (Elamipretide) | Normalized the increase in proton leak and reduced mitochondrial ROS in cardiomyocytes via targeting Cardiolipin | [91] |
| Inflammation | β-hydroxybutyrate | Activation of CS (citrate synthase) and inhibition of fatty acid uptake | [93] |
| Oxidative stress | Antioxidant enzyme catalase | Alleviate mitochondrial oxidative stress | [92] |
| Metabolic imbalance | Acetylcarnitine | Improved aging-induced decreases in OXPHOS, complex III and complex IV | [6] |
Metabolites and dietary supplements for pharmacological interventions
NAD+ repletion can delay several hallmarks of aging and suppress the deterioration of age-related diseases[95]. This suggests a significant potential for the treatment of cardiac diseases in the elderly population with supplementation of NAD+ precursors. Indeed, the dietary intake of NAM could reduce cardiac hypertrophy and diastolic dysfunction in aged mice[96] [Figure 5]. However, challenges still exist. Several preclinical studies have confirmed that both NA and NAM treatment can cause side effects such as painful flushing sensations[97-99]. The effect of NMN treatment was tested on old mouse hearts[100], and supplementing this metabolite could restore mitochondria and heart function [Table 2]. However, some potential side effects of NMN have also been proposed, especially concomitant with high-dose administration, such as hepatic pressure and cancer growth[101]. Additionally, NR may be a more suitable NAD+ precursor, since it was not found to be associated with flushing or other severe side effects[102]. Oral administration of NR has been shown to increase NAD+ levels in humans[103]. Moreover, NR can prevent the deterioration of cardiac function and adverse remodeling in a mouse model of dilated cardiomyopathy[104]. However, NR is unstable in blood circulation due to degradation to NAM, thus reducing its availability in the heart after oral supplementation[105]. In addition, the therapeutic value of NR still has certain limitations regarding its production methods, including low yield and the use of expensive or hazardous reagents[106]. In summary, to adapt NR or NMN treatment for therapeutic usage against cardiac aging, it is required to determine oral availability and therapeutic dosage.

Figure 5. Pharmacological interventions against cardiac aging. NR: Nicotinamide riboside; NAM: nicotinamide; NMN: nicotinamide mononucleotide; NAMPT: nicotinamide phosphoribosyltransferase; AMPK: AMP-activated protein kinase; βOHB: β-hydroxybutyrate; SS-31: elamipretide; CL: cardiolipin; SP: spermine; SPD: spermidine; GABR: global arginine bioavailability ratio; CS: Citrate synthase; ROS: reactive oxygen species.
Pharmacological interventions for cardiac rejuvenation
| Therapeutic strategies | Target | Effect | Reference |
| NMN | NAD+ | Fully reversed an age-related deficiency in systolic function | [100] |
| β-hydroxybutyrate | Fatty acid uptake | Antagonize proinflammatory cytokine-triggered mitochondrial dysfunction | [93] |
| Spermine | Lipid metabolism and glutathione metabolism pathways | Inhibited age-related myocardial fibrosis and cell apoptosis | [110] |
| Spermidine | Mitochondrial respiration and titin phosphorylation | Reduced blood pressure and delayed the progression to heart failure | [111] |
| Uridine | Metabolic process and inflammation | Improved the function of the heart | [107] |
| Oleate | Increased the levels of NAD+ | Increased anti-aging metabolites | [109] |
| NAD+ | Metabolic process | Improved the function of the heart | [120] |
| Curcumin | Activation of AMPK | Inhibited age-related oxidative changes | [121] |
| Resveratrol | SIRT1 | Ameliorated aging-related metabolic phenotypes | [113] |
| SRT1720 | SIRT1 | Reduced age-related loss of heart function | [114] |
A recent study identified uridine, a pyrimidine nucleoside, as a metabolite that can rejuvenate aged human stem cells and promote the regeneration of various tissues, including the heart[107]. Interestingly, uridine was proved to have an anti-inflammatory effect via modulating inhibitor of kappa B kinaseα/β(IKKα/β) and nuclear factor-kappaB (NF-κB) signaling[108]. This anti-inflammatory effect of uridine may provide a more amiable environment for aging cardiomyocytes. Oleate, an unsaturated fatty acid, could increase anti-aging metabolites such as NAD+ levels in vivo[109]. Polyamines such as spermine (SP) are essential for cell growth, and their levels decline with age. Interestingly, SP treatment could reverse and inhibit age-related myocardial morphology alterations and apoptosis[110]. SP treatment upregulates the expression of pyruvate kinase M1/2 (PKM), enolase 3 (ENO3), and phosphoglycerate mutase 2 (PGAM2), therefore enhancing cardiac lipid metabolism. SP treatment also downregulates glutathione S-transferase alpha 3 (GSTA3) and dehydroascorbic acid production, inhibiting glutathione metabolism and protecting against cardiac aging[110] [Figure 5]. Polyamine spermidine (SPD) also has cardioprotective effects[110,111]. SPD treatment could reduce cardiac hypertrophy and preserve diastolic function in old mice[111]. This protective effect was achieved by improving the global arginine bioavailability ratio (GABR), which favors the production of nitric oxide (NO) and subsequently decreases systemic blood pressure[111]. SPD treatment can also increase titin phosphorylation and improve the mechanical properties of cardiomyocytes[111]. In summary, these cardioprotective metabolites could serve as potential clinical therapeutics that target cardiac aging.
Dietary supplements offer a convenient resource for restoring cardiac youthfulness in the aging population [Table 2]. Among them, several naturally occurring molecules targeting longevity pathways could improve mitochondrial physiology. For example, resveratrol has been proven to enhance mitochondrial biogenesis in aging mice[112]. Mechanically, resveratrol activates the cyclic adenosine monophosphate (cAMP)/exchange protein directly activated by the cAMP 1 (Epac1)/AMPK pathway, which subsequently increases the NAD+ level and the activity of SIRT1[113]. SRT1720, another compound activating SIRT1, has health and lifespan benefits in adult mice[114]. Thymoquinone and curcumin effectively suppressed the aging-associated oxidative alterations in mice hearts[115]. Curcumin could also improve cardiac angiogenesis and promote heart performance in senescent rats[116]. Curcumin could activate AMPK signaling, thereby promoting autophagy and alleviating cardiac apoptosis[117].
Interestingly, a combination of different anti-aging agents may achieve better cardiac rejuvenation. Two different mitochondrially targeted drugs, SS-31 and NMN, were tested on old mouse hearts[100]. Combining them resulted in a synergistic effect on old hearts that best recapitulated the young state. Moreover, a synergistic effect of leucine-resveratrol combinations on glucose homeostasis and insulin sensitivity was observed in patients with prediabetes[118,119]. These cardiac anti-aging strategies are gaining popularity, and optimizing the drug combination or targeting will undoubtedly facilitate the development of anti-aging therapies. Moreover, further mechanistic studies are needed for drug safety and efficacy assessment of cardiac anti-aging strategies.
CONCLUSIONS
CVDs associated with aging are the leading global healthcare burden in the 21st century. Research focusing on metabolic dysfunction in the aging process might identify novel specific agents. Interestingly, the driving factors of cardiac aging influence each other; thus, strategies targeting multiple driving factors may have a synergistic effect. This review examines metabolic components involved in cardiac aging and how they influence the main aging features. The modulation of these components and correlative pathways could improve human cardiac health and prevent major age-related CVDs. Maintaining healthy mitochondria and metabolic regulation is essential to long-term cardiac health. This review also summarizes different approaches to reversing metabolic changes in cardiac aging. It is challenging to only focus on specific cardiac pathologies because of the multi-organ involvement in age-associated CVDs. Therefore, new agents targeting communications between multiple organs could pave the way to understanding the complex nature of CVDs in the aged population. In conclusion, a thorough understanding of the role of metabolic regulation in human cardiac aging will be needed to combat age-related CVDs.
DECLARATIONS
Authors’ contributionsParticipated in research design: Xiao J, Vulugundam G
Performed data analysis: Zhang X, Hu M, Lu Y
Wrote or contributed to the writing of the manuscript: Liu C, Gokulnath P
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the grants from National Key Research and Development Project (2018YFE0113500 to Xiao J), National Natural Science Foundation of China (82020108002 and 81911540486 to Xiao J), Innovation Program of Shanghai Municipal Education Commission (2017-01-07-00-09-E00042 to Xiao J), the grant from Science and Technology Commission of Shanghai Municipality (20DZ2255400 and 21XD1421300 to Xiao J), the “Dawn” Program of Shanghai Education Commission (19SG34 to Xiao J), and National Natural Science Foundation of China (82000287 to Liu C).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Moturi S, Ghosh-Choudhary SK, Finkel T. Cardiovascular disease and the biology of aging. J Mol Cell Cardiol 2022;167:109-17.
2. Yazdanyar A, Newman AB. The burden of cardiovascular disease in the elderly: morbidity, mortality, and costs. Clin Geriatr Med 2009;25:563-77,vii.
3. Gude NA, Broughton KM, Firouzi F, Sussman MA. Cardiac ageing: extrinsic and intrinsic factors in cellular renewal and senescence. Nat Rev Cardiol 2018;15:523-42.
4. Borlaug BA, Olson TP, Lam CS, et al. Global cardiovascular reserve dysfunction in heart failure with preserved ejection fraction. J Am Coll Cardiol 2010;56:845-54.
5. Li H, Hastings MH, Rhee J, Trager LE, Roh JD, Rosenzweig A. Targeting age-related pathways in heart failure. Circ Res 2020;126:533-51.
6. Lesnefsky EJ, Chen Q, Hoppel CL. Mitochondrial metabolism in aging heart. Circ Res 2016;118:1593-611.
7. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013;153:1194-217.
8. Picca A, Mankowski RT, Burman JL, et al. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat Rev Cardiol 2018;15:543-54.
9. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol 2018;28:436-53.
10. Sahin E, Colla S, Liesa M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011;470:359-65.
11. Marzetti E, Wohlgemuth SE, Anton SD, Bernabei R, Carter CS, Leeuwenburgh C. Cellular mechanisms of cardioprotection by calorie restriction: state of the science and future perspectives. Clin Geriatr Med 2009;25:715-32, ix.
12. HARMAN D. Aging: a theory based on free radical and radiation chemistry. J Gerontol 1956;11:298-300.
13. Duicu OM, Mirica SN, Gheorgheosu DE, Privistirescu AI, Fira-Mladinescu O, Muntean DM. Ageing-induced decrease in cardiac mitochondrial function in healthy rats. Can J Physiol Pharmacol 2013;91:593-600.
14. Phaneuf S, Leeuwenburgh C. Cytochrome c release from mitochondria in the aging heart: a possible mechanism for apoptosis with age. Am J Physiol Regul Integr Comp Physiol 2002;282:R423-30.
15. Logan A, Shabalina IG, Prime TA, et al. In vivo levels of mitochondrial hydrogen peroxide increase with age in mtDNA mutator mice. Aging Cell 2014;13:765-8.
16. Tang X, Li PH, Chen HZ. Cardiomyocyte senescence and cellular communications within myocardial microenvironments. Front Endocrinol (Lausanne) 2020;11:280.
17. Shah MS, Brownlee M. Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circ Res 2016;118:1808-29.
18. Himelman E, Lillo MA, Nouet J, et al. Prevention of connexin-43 remodeling protects against Duchenne muscular dystrophy cardiomyopathy. J Clin Invest 2020;130:1713-27.
19. Segovia-Roldan M, Diez ER, Pueyo E. Melatonin to rescue the aged heart: antiarrhythmic and antioxidant benefits. Oxid Med Cell Longev 2021;2021:8876792.
20. Makino N, Maeda T, Oyama J, et al. Antioxidant therapy attenuates myocardial telomerase activity reduction in superoxide dismutase-deficient mice. J Mol Cell Cardiol 2011;50:670-7.
21. Calvani M, Reda E, Arrigoni-Martelli E. Regulation by carnitine of myocardial fatty acid and carbohydrate metabolism under normal and pathological conditions. Basic Res Cardiol 2000;95:75-83.
22. Moreau R, Heath SH, Doneanu CE, Harris RA, Hagen TM. Age-related compensatory activation of pyruvate dehydrogenase complex in rat heart. Biochem Biophys Res Commun 2004;325:48-58.
23. Kolwicz SC Jr, Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res 2012;111:728-38.
24. Ritterhoff J, Young S, Villet O, et al. Metabolic remodeling promotes cardiac hypertrophy by directing glucose to aspartate biosynthesis. Circ Res 2020;126:182-96.
25. Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 1996;94:2837-42.
26. Karbowska J, Kochan Z, Smolenski RT. Peroxisome proliferator-activated receptor alpha is downregulated in the failing human heart. Cell Mol Biol Lett ;2003:8:49-53.
27. Abdellatif M, Sedej S, Kroemer G. NAD+ metabolism in cardiac health, aging, and disease. Circulation 2021;144:1795-817.
28. Nikiforov A, Dölle C, Niere M, Ziegler M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J Biol Chem 2011;286:21767-78.
29. Kropotov A, Kulikova V, Nerinovski K, et al. Equilibrative nucleoside transporters mediate the import of nicotinamide riboside and nicotinic acid riboside into human cells. Int J Mol Sci 2021;22:1391.
30. Grozio A, Mills KF, Yoshino J, et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat Metab 2019;1:47-57.
31. Luongo TS, Eller JM, Lu MJ, et al. SLC25A51 is a mammalian mitochondrial NAD+ transporter. Nature 2020;588:174-9.
33. Yoshida M, Satoh A, Lin JB, et al. Extracellular vesicle-contained eNAMPT delays aging and extends lifespan in mice. Cell Metab 2019;30:329-342.e5.
34. Camacho-Pereira J, Tarragó MG, Chini CCS, et al. CD38 dictates age-related NAD Decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab 2016;23:1127-39.
35. Covarrubias AJ, Kale A, Perrone R, et al. Senescent cells promote tissue NAD+ decline during ageing via the activation of CD38+ macrophages. Nat Metab 2020;2:1265-83.
36. Tarragó MG, Chini CCS, Kanamori KS, et al. A potent and specific CD38 Inhibitor ameliorates age-related metabolic dysfunction by reversing tissue NAD+ decline. Cell Metab 2018;27:1081-1095.e10.
37. Malavasi F, Deaglio S, Funaro A, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev 2008;88:841-86.
38. Chini CCS, Peclat TR, Warner GM, et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD+ and NMN levels. Nat Metab 2020;2:1284-304.
39. Chini CCS, Tarragó MG, Chini EN. NAD and the aging process: role in life, death and everything in between. Mol Cell Endocrinol 2017;455:62-74.
40. Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One 2011;6:e19194.
41. Covarrubias AJ, Perrone R, Grozio A, Verdin E. NAD+ metabolism and its roles in cellular processes during ageing. Nat Rev Mol Cell Biol 2021;22:119-41.
42. Lee JS, Park AH, Lee SH, et al. Beta-lapachone, a modulator of NAD metabolism, prevents health declines in aged mice. PLoS One 2012;7:e47122.
43. Morales J, Li L, Fattah FJ, et al. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit Rev Eukaryot Gene Expr 2014;24:15-28.
45. Du J, Zhou Y, Su X, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011;334:806-9.
46. Tan M, Peng C, Anderson KA, et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab 2014;19:605-17.
47. Hirschey MD, Shimazu T, Goetzman E, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010;464:121-5.
48. Jeong SM, Xiao C, Finley LW, et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell 2013;23:450-63.
49. Sinclair DA, Guarente L. Small-molecule allosteric activators of sirtuins. Annu Rev Pharmacol Toxicol 2014;54:363-80.
52. Winnik S, Auwerx J, Sinclair DA, Matter CM. Protective effects of sirtuins in cardiovascular diseases: from bench to bedside. Eur Heart J 2015;36:3404-12.
53. Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 2004;23:2369-80.
54. Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem 2008;283:27628-35.
55. Sugden MC, Caton PW, Holness MJ. PPAR control: it’s SIRTainly as easy as PGC. J Endocrinol 2010;204:93-104.
56. Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 2011;93:884S-90.
57. Sadhukhan S, Liu X, Ryu D, et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc Natl Acad Sci USA 2016;113:4320-5.
58. Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature 2013;497:217-23.
59. Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal 2009;2:ra75.
60. Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol 2020;21:183-203.
61. Kaeberlein M, Powers RW 3rd, Steffen KK, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005;310:1193-6.
62. Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008;320:1496-501.
63. Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008;30:214-26.
64. Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol 2018;19:579-93.
65. Yan M, Sun S, Xu K, et al. Cardiac aging: from basic research to therapeutics. Oxid Med Cell Longev 2021;2021:9570325.
66. Tang X, Chen XF, Wang NY, et al. SIRT2 acts as a cardioprotective deacetylase in pathological cardiac hypertrophy. Circulation 2017;136:2051-67.
67. Dadson K, Chasiotis H, Wannaiampikul S, Tungtrongchitr R, Xu A, Sweeney G. Adiponectin mediated APPL1-AMPK signaling induces cell migration, MMP activation, and collagen remodeling in cardiac fibroblasts. J Cell Biochem 2014;115:785-93.
68. Cieslik KA, Taffet GE, Crawford JR, Trial J, Mejia Osuna P, Entman ML. AICAR-dependent AMPK activation improves scar formation in the aged heart in a murine model of reperfused myocardial infarction. J Mol Cell Cardiol 2013;63:26-36.
69. Steinberg GR, Schertzer JD. AMPK promotes macrophage fatty acid oxidative metabolism to mitigate inflammation: implications for diabetes and cardiovascular disease. Immunol Cell Biol 2014;92:340-5.
70. Zhang R, Chen HZ, Liu JJ, et al. SIRT1 suppresses activator protein-1 transcriptional activity and cyclooxygenase-2 expression in macrophages. J Biol Chem 2010;285:7097-110.
71. Pearce EL, Walsh MC, Cejas PJ, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009;460:103-7.
72. Tang X, Luo YX, Chen HZ, Liu DP. Mitochondria, endothelial cell function, and vascular diseases. Front Physiol 2014;5:175.
73. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 2018;19:121-35.
74. Zhang W, Wang Q, Wu Y, et al. Endothelial cell-specific liver kinase B1 deletion causes endothelial dysfunction and hypertension in mice in vivo. Circulation 2014;129:1428-39.
75. Hirschi KK, Ingram DA, Yoder MC. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler Thromb Vasc Biol 2008;28:1584-95.
76. Wang C, Wang F, Li Z, Huang L, Cao Q, Chen S. MeCP2 mediated dysfunction in senescent EPCs. Oncotarget 2017;8:78289-99.
77. Zha S, Li Z, Cao Q, Wang F, Liu F. PARP1 inhibitor (PJ34) improves the function of aging-induced endothelial progenitor cells by preserving intracellular NAD+ levels and increasing SIRT1 activity. Stem Cell Res Ther 2018;9:224.
78. Bots SH, Peters SAE, Woodward M. Sex differences in coronary heart disease and stroke mortality: a global assessment of the effect of ageing between 1980 and 2010. BMJ Glob Health 2017;2:e000298.
79. Ghali JK, Piña IL, Gottlieb SS, Deedwania PC, Wikstrand JC. Metoprolol CR/XL in female patients with heart failure: analysis of the experience in Metoprolol Extended-Release Randomized Intervention Trial in Heart Failure (MERIT-HF). Circulation 2002;105:1585-91.
80. Pang L, Jiang X, Lian X, et al. Caloric restriction-mimetics for the reduction of heart failure risk in aging heart: with consideration of gender-related differences. Mil Med Res 2022;9:33.
81. de Arellano ML, Pozdniakova S, Kühl AA, Baczko I, Ladilov Y, Regitz-Zagrosek V. Sex differences in the aging human heart: decreased sirtuins, pro-inflammatory shift and reduced anti-oxidative defense. Aging (Albany NY) 2019;11:1918-33.
82. Widder J, Pelzer T, von Poser-Klein C, et al. Improvement of endothelial dysfunction by selective estrogen receptor-alpha stimulation in ovariectomized SHR. Hypertension 2003;42:991-6.
83. Konhilas JP, Leinwand LA. The effects of biological sex and diet on the development of heart failure. Circulation 2007;116:2747-59.
84. Liu Z, Gou Y, Zhang H, et al. Estradiol improves cardiovascular function through up-regulation of SOD2 on vascular wall. Redox Biol 2014;3:88-99.
85. Arad M, Seidman CE, Seidman JG. AMP-activated protein kinase in the heart: role during health and disease. Circ Res 2007;100:474-88.
86. Feng W, Liu J, Wang S, et al. Alginate oligosaccharide alleviates D-galactose-induced cardiac ageing via regulating myocardial mitochondria function and integrity in mice. J Cell Mol Med 2021;25:7157-68.
87. James AM, Sharpley MS, Manas AR, et al. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J Biol Chem 2007;282:14708-18.
88. Mao G, Kraus GA, Kim I, et al. A mitochondria-targeted vitamin E derivative decreases hepatic oxidative stress and inhibits fat deposition in mice. J Nutr 2010;140:1425-31.
89. Tao R, Xiong X, DePinho RA, Deng CX, Dong XC. FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. J Biol Chem 2013;288:29252-9.
90. Vasan RS, Xanthakis V, Lyass A, et al. Epidemiology of left ventricular systolic dysfunction and heart failure in the framingham study: an echocardiographic study over 3 decades. JACC Cardiovasc Imaging 2018;11:1-11.
91. Chiao YA, Zhang H, Sweetwyne M, et al. Late-life restoration of mitochondrial function reverses cardiac dysfunction in old mice. Elife 2020;9:e55513.
92. Dai DF, Chen T, Wanagat J, et al. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 2010;9:536-44.
93. Deng Y, Xie M, Li Q, et al. Targeting mitochondria-inflammation circuit by β-hydroxybutyrate mitigates HFpEF. Circ Res 2021;128:232-45.
94. Costantino S, Paneni F, Cosentino F. Ageing, metabolism and cardiovascular disease. J Physiol 2016;594:2061-73.
95. Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J Biol Chem 2005;280:43121-30.
96. Abdellatif M, Trummer-Herbst V, Koser F, et al. Nicotinamide for the treatment of heart failure with preserved ejection fraction. Sci Transl Med 2021;13:eabd7064.
97. Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr 2008;28:115-30.
98. Reiten OK, Wilvang MA, Mitchell SJ, Hu Z, Fang EF. Preclinical and clinical evidence of NAD+ precursors in health, disease, and ageing. Mech Ageing Dev 2021;199:111567.
99. MacKay D, Hathcock J, Guarneri E. Niacin: chemical forms, bioavailability, and health effects. Nutr Rev 2012;70:357-66.
100. Whitson JA, Bitto A, Zhang H, et al. SS-31 and NMN: two paths to improve metabolism and function in aged hearts. Aging Cell 2020;19:e13213.
101. Yoshino J, Baur JA, Imai SI. NAD+ intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab 2018;27:513-28.
102. Martens CR, Denman BA, Mazzo MR, et al. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults. Nat Commun 2018;9:1286.
103. Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a preiss-handler independent route to NAD+ in fungi and humans. Cell 2004;117:495-502.
104. Diguet N, Trammell SAJ, Tannous C, et al. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation 2018;137:2256-73.
105. Ratajczak J, Joffraud M, Trammell SA, et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat Commun 2016;7:13103.
106. Mehmel M, Jovanović N, Spitz U. Nicotinamide riboside-the current state of research and therapeutic uses. Nutrients 2020;12:1616.
107. Liu Z, Li W, Geng L, et al. Cross-species metabolomic analysis identifies uridine as a potent regeneration promoting factor. Cell Discov 2022;8:6.
108. Jeengar MK, Thummuri D, Magnusson M, Naidu VGM, Uppugunduri S. Uridine ameliorates dextran sulfate sodium (DSS)-induced colitis in mice. Sci Rep 2017;7:3924.
109. Enot DP, Niso-Santano M, Durand S, et al. Metabolomic analyses reveal that anti-aging metabolites are depleted by palmitate but increased by oleate in vivo. Cell Cycle 2015;14:2399-407.
110. Zhang H, Wang J, Li L, et al. Spermine and spermidine reversed age-related cardiac deterioration in rats. Oncotarget 2017;8:64793-808.
111. Eisenberg T, Abdellatif M, Schroeder S, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med 2016;22:1428-38.
112. Baur JA, Pearson KJ, Price NL, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006;444:337-42.
113. Park SJ, Ahmad F, Philp A, et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012;148:421-33.
114. Mitchell SJ, Martin-Montalvo A, Mercken EM, et al. The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep 2014;6:836-43.
115. El-Far AH, Elewa YHA, Abdelfattah EA, et al. Thymoquinone and curcumin defeat aging-associated oxidative alterations induced by D-galactose in rats’ brain and heart. Int J Mol Sci 2021;22:6839.
116. Ghorbanzadeh V, Pourheydar B, Dariushnejad H, Ghalibafsabbaghi A, Chodari L. Curcumin improves angiogenesis in the heart of aged rats: involvement of TSP1/NF-κB/VEGF-A signaling. Microvasc Res 2022;139:104258.
117. Yao Q, Ke ZQ, Guo S, et al. Curcumin protects against diabetic cardiomyopathy by promoting autophagy and alleviating apoptosis. J Mol Cell Cardiol 2018;124:26-34.
118. Amorim JA, Coppotelli G, Rolo AP, Palmeira CM, Ross JM, Sinclair DA. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat Rev Endocrinol 2022;18:243-58.
119. Zemel MB. Modulation of energy sensing by leucine synergy with natural sirtuin activators: effects on health span. J Med Food 2020;23:1129-35.
120. Hershberger KA, Martin AS, Hirschey MD. Role of NAD+ and mitochondrial sirtuins in cardiac and renal diseases. Nat Rev Nephrol 2017;13:213-25.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Liu C, Zhang X, Hu M, Lu Y, Gokulnath P, Vulugundam G, Xiao J. Metabolic targets in cardiac aging and rejuvenation. J Cardiovasc Aging 2022;2:46. http://dx.doi.org/10.20517/jca.2022.31
AMA Style
Liu C, Zhang X, Hu M, Lu Y, Gokulnath P, Vulugundam G, Xiao J. Metabolic targets in cardiac aging and rejuvenation. The Journal of Cardiovascular Aging. 2022; 2(4): 46. http://dx.doi.org/10.20517/jca.2022.31
Chicago/Turabian Style
Liu, Chang, Xiao Zhang, Meiyu Hu, Yi Lu, Priyanka Gokulnath, Gururaja Vulugundam, Junjie Xiao. 2022. "Metabolic targets in cardiac aging and rejuvenation" The Journal of Cardiovascular Aging. 2, no.4: 46. http://dx.doi.org/10.20517/jca.2022.31
ACS Style
Liu, C.; Zhang X.; Hu M.; Lu Y.; Gokulnath P.; Vulugundam G.; Xiao J. Metabolic targets in cardiac aging and rejuvenation. J. Cardiovasc. Aging. 2022, 2, 46. http://dx.doi.org/10.20517/jca.2022.31
About This Article
Copyright

Data & Comments
Data


 Cite This Article 16 clicks
Cite This Article 16 clicks

 Like This Article 23
likes
Like This Article 23
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.