
A role for somatic mutations in the evolution of lifespan
 0
0 ,
, The spectrum of lifespans across the animal kingdom ranges from days to hundreds of years, with a few species that even display biological immortality. For mammals, lifespans of about a year in rodents (Müller’s giant Sunda rat) to more than 200 years in the bowhead whale are observed. This variety across evolution and its relation to organismal, cellular, and molecular aging processes constitutes one of the longest-standing mysteries in biology. Over time many theories have been put forward to explain this lifespan range, but only few definitive answers have been found so far. Early models proposed that an accumulation of mutations in somatic tissues leads to a gradual buildup of faulty proteins and mistakes in gene regulatory regions, ultimately impairing cell fitness and function and consequentially damaging tissues and organs[1]. However, recent evidence demonstrated that mutation rates, for example, in humans, are much lower than required for gene function loss in a meaningful number of cells and, additionally, that somatic cells very well tolerate the linear mutation accumulation that occurs throughout life[2]. These insights very strongly suggest that somatic mutations do not directly impair cell proliferation or function and therefore should have a minor, if any, impact on lifespan.
Despite this history, the somatic mutation theory is still considered relevant today due to another phenomenon observed throughout the age: clonal expansion. Mutated cells can undergo positive selection and takeover substantial fractions of the overall cell population in many, presumably all tissues that have a turnover much smaller than the species’ lifespan. In humans, these are almost all tissues with a few exceptions, most notably muscle, and neural tissues.
Small advantages in the proliferation of individual cells occur often, and clonal expansion is therefore observed frequently even without giving rise to cancer, its most extreme manifestation. In non-cancerous clonal expansion, cell clones outcompete their peers and thereby gradually aggregate in the tissue; importantly, this happens irrespective of their physiological function. Therefore, clones can expand that are detrimental to the tissue and eventually to organismal function[3]. This colonization of tissues can shrink the pool of genetic makeups and cellular phenotypes and thereby limit the options for stress response, rendering the organism increasingly fragile. A prominent example is the clonal hematopoiesis of indeterminate potential, which seems to increase the risk of age-related cardiovascular diseases[4].
So far, rigorous testing of the role of somatic mutation rates in disease and lifespan was very difficult due to inter-cell variability of genomes, the resulting need for in vitro expansion of cell clones and technical limitations of whole-genome DNA sequencing itself.
In a recent study, Cagan et al. took on this challenge and explored somatic mutation rates as one potentially important factor in aging and in controlling maximal lifespan[5]. Using laser-capture microdissection on healthy intestinal crypts of 16 mammals and subjecting the samples to low-input DNA sequencing, the authors were able to measure mutation rates and correlate them with maximum lifespans observed in captivity, i.e., in the absence of extrinsic mortality. Intestinal crypts are clonal as they emerge from individual stem cells and their mutations are thought to largely arise from endogenous rather than environmental stressors from which a crypt is shielded. Also, mutations in crypts accumulate linearly over the lifetime, which makes them an attractive, interpretable experimental system. Different forms of DNA damage and the subsequent DNA repair response are associated with different frequencies and types of mutations, i.e., mutational signatures. Strikingly, the authors found that the same three signatures dominate the mutations recorded in the colons across all investigated species. This result argues that the approach indeed can be used to investigate somatic mutation rates across evolution.
This cross-species mutation analysis revealed a striking anticorrelation with lifespan: the longer the life of a given species, the smaller the somatic mutation rate per year.
With the reasonable assumptions that mutagenic processes are similar across an individual’s life and across crypts of different species and by testing potentially confounding life-history traits like the basal metabolic rate, litter size, or body mass, the authors conclude that the variation in mutation rates is mostly explained by lifespan. For example, mice display a 17-fold higher somatic mutation rate than humans, a difference too big to be explained by differences in cell division rate or other confounding factors. This suggests that somatic mutations contribute to the forty-fold difference in maximal lifespan and that lifespan and potentially aging too are indeed evolutionary constrained[5].
The observations also suggest a solution to the following paradox: simple modeling of carcinogenesis predicts a positive correlation of cancer risk with cell number, i.e., with body mass and lifespan. This, however, is not observed: large, long-lived animals found a way to suppress cancer formation that is much more efficient than that of small, short-lived animals, which results in similar cancer rates across species (Peto’s paradox). In the study at hand, the mutational load, when calculated as the end-of-lifespan burden per cell, was relatively similar across species (3-fold variation) relative to the close to forty-thousand-fold variation in body mass, strongly arguing for effective mutation control in long-lived, big animals. This may be achieved through selection on the various DNA repair pathways or by ways of clearance of tumorigenic cells.
It is tempting to speculate that other age-associated functional deterioration of biomolecules, cells, and tissues, such as the loss of resilience or regenerative capacity, could be suppressed in an analogous way [Figure 1].
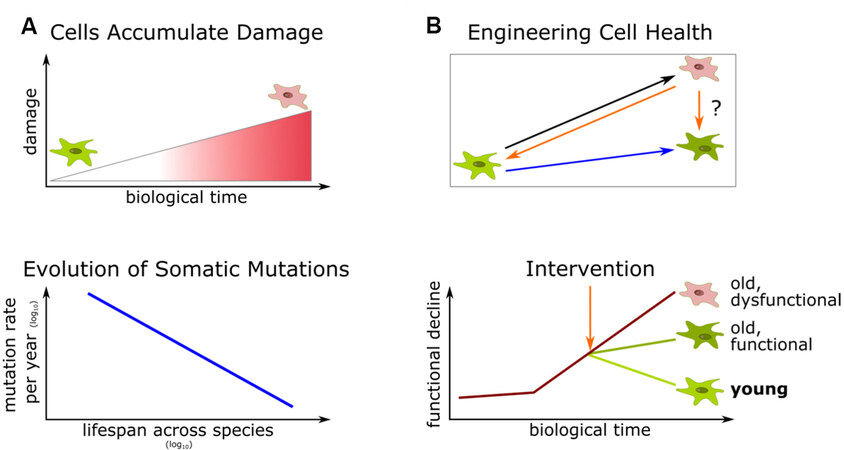
Figure 1. The evolution of somatic mutation rates suggests that cell health is amenable to engineering by exploiting mechanisms naturally present in long-lived species. (A) Cells gradually accumulate different types of molecular damage, including DNA mutations, as they progress through biological time, eventually resulting in old dysfunctional cells (red). Cagan et al. report differences in somatic mutation rates across species, suggesting that this damage is under evolutionary constraint[5]. (B) This observation shows that this and potentially other age-related losses of function are in principle amenable to interventions that recapitulate the natural molecular processes in long-lived animals. Such interventions could program cell trajectories to either halt decline at a tolerable level (dark green cell) or reverse aging phenotypes (bright green cell).
A great example of how technological advances allow tackling old problems, this work raises many immediate questions. If somatic mutations play an important role in aging and lifespan, how can this be reconciled with the observation that mice and humans with germline mutations in DNA polymerases or repair enzymes and a resulting increase of mutations, do not display premature aging? Also, mammals cloned from old and therefore mutated somatic cells produce offspring with a normal lifespan, which, although affected by survivorship bias, argues against a prime role of mutations in aging[6].
One explanation could be that the complex nature of aging limits the impact of any individual contributing factor (here, mutation rate) and, moreover, that this impact is non-linearly affecting a small subset of age-related phenotypes only. On the other hand, improved DNA repair is reported for long-lived species and enzyme variants of this pathway are even enriched in human super-centenarians, which supports the authors’ model[7]. Another complication arises from a recent observation in plants where a non-random, constrained mutation frequency for gene bodies and essential genes was found. Currently, it is believed that this phenomenon is mediated by physical and epigenomic features of these regions[8]. Assuming that this mutation bias is present in animals too, it would be very interesting to analyze mutation rates across evolution with resolution of the functional units of the genome.
Any potential link to the epigenome bears interesting implications since the DNA damage response evokes transient chromatin remodeling, its enzymes are themselves under epigenetic control and loss of epigenetic regulation is a main feature of aging. Aberrant expression of stem cell transcription factors can be used to remodel the epigenome and reverse cellular and organismal hallmarks of aging. This includes inducing regenerative capacity in adult cardiomyocytes, a feature that previously was thought to be limited to fetal hearts only[9]. These experiments suggest a novel therapeutic path with enormous clinical potential for the cardiovascular field. In such experimental cellular programming settings, potential effects on mutation rates and clonal expansion recorded by lineage tracing are likely to elucidate, clinically relevant aging mechanisms. For future studies, it will be important to identify and probe non-mutation related anti-stress mechanisms that show divergence across species and test if, for example, the outstanding regenerative capacity a few animals display can be experimentally recapitulated in murine and human laboratory systems. We deem the study of molecular means of regeneration and cellular resilience in homeostasis as particularly relevant for human health.
The study by Cagan et al. provides an exciting example of how seemingly unchangeable processes of decay indeed can be modulated[5]. The causes of human aging and the many debilitating age-related effects including the high incidence of cardiovascular diseases are manifold. However, the recent rapid expansion of the lifespan of our species, unprecedented in the history of the planet, is certainly one of them. Manipulating the genetic and epigenetic machinery to counteract the negative side-effects of this step of human development is one of the most worthwhile challenges in science.
DECLARATIONS
Authors’ contributionsConceived and wrote the paper: Memczak S, Izpisua Belmonte JC
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestThe authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
2. Vijg J, Dong X. Pathogenic mechanisms of somatic mutation and genome mosaicism in aging. Cell 2020;182:12-23.
4. Jaiswal S, Libby P. Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat Rev Cardiol 2020;17:137-44.
5. Cagan A, Baez-Ortega A, Brzozowska N, et al. Somatic mutation rates scale with lifespan across mammals. Nature 2022;604:517-24.
7. Garagnani P, Marquis J, Delledonne M, et al. Whole-genome sequencing analysis of semi-supercentenarians. Elife 2021;10:e57849.
8. Monroe JG, Srikant T, Carbonell-Bejerano P, et al. Mutation bias reflects natural selection in Arabidopsis thaliana. Nature 2022;602:101-5.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Memczak S, Izpisua Belmonte JC. A role for somatic mutations in the evolution of lifespan. J Cardiovasc Aging 2022;2:39. http://dx.doi.org/10.20517/jca.2022.28
AMA Style
Memczak S, Izpisua Belmonte JC. A role for somatic mutations in the evolution of lifespan. The Journal of Cardiovascular Aging. 2022; 2(4): 39. http://dx.doi.org/10.20517/jca.2022.28
Chicago/Turabian Style
Memczak, Sebastian, Juan Carlos Izpisua Belmonte. 2022. "A role for somatic mutations in the evolution of lifespan" The Journal of Cardiovascular Aging. 2, no.4: 39. http://dx.doi.org/10.20517/jca.2022.28
ACS Style
Memczak, S.; Izpisua Belmonte JC. A role for somatic mutations in the evolution of lifespan. J. Cardiovasc. Aging. 2022, 2, 39. http://dx.doi.org/10.20517/jca.2022.28
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 6 clicks
Cite This Article 6 clicks

 Like This Article 1
likes
Like This Article 1
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.