
Gut microbiota in sarcopenia and heart failure


Abstract
Sarcopenia is common in aging and in patients with heart failure (HF) who may experience worse outcomes. Patients with muscle wasting are more likely to experience falls and can have serious complications when undergoing cardiac procedures. While intensive nutritional support and exercise rehabilitation can help reverse some of these changes, they are often under-prescribed in a timely manner, and we have limited insights into who would benefit. Mechanistic links between gut microbial metabolites (GMM) have been identified and may contribute to adverse clinical outcomes in patients with cardio-renal diseases and aging. This review will examine the emerging evidence for the influence of the gut microbiome-derived metabolites and notable signaling pathways involved in both sarcopenia and HF, especially those linked to dietary intake and mitochondrial metabolism. This provides a unique opportunity to gain mechanistic and clinical insights into developing novel therapeutic strategies that target these GMM pathways or through tailored nutritional modulation to prevent progressive muscle wasting in elderly patients with heart failure.
Keywords
INTRODUCTION
Heart failure (HF), a complex clinical syndrome that results from various types of heart disease, is the top killer of the human population, taking an estimated 17.9 million lives each year[1]. It is estimated that
The term sarcopenia, was initially used by Rosenberg in 1989 as an age-dependent condition of the loss of muscle mass and function[6]. The current updated definition of sarcopenia is now considered as a muscle disease rooted in decrease of muscle strength, reduction of muscle quantity and quality (muscle wasting) and decline of physical performance according to the European Working Group on Sarcopenia in Older People[7]. Sarcopenia is common among older adults with a prevalence between 10% and 40% but can also occur earlier in life when associated with systemic disease linked to inflammatory processes, e.g., malignancy or organ failure[7]. It was shown that the prevalence of sarcopenia in patients with HF was nearly 20% higher than the control subjects of the same age[8-11]. Mechanistically, HF and sarcopenia may share common determinants such as inflammation, hormonal alterations, malnutrition, oxidative stress and mitochondria dysfunction[12,13].
Recent interests have focused on gut microbiota in human diseases and healthy aging. The human gut microbiota consists of a huge number of microorganisms. Among them, anaerobic Bacteroidetes and Firmicutes contribute > 90% of the total bacterial species in healthy guts. Perturbation of the gut microbiota or dysbiosis contributes to various diseases, including cardiovascular diseases (CVD), muscle wasting, and aging[14]. Understanding the interplay between gut microbiota and heart/skeletal muscle may help us to better identify vulnerability and investigate targeted interventions.
This review focuses on recent advances in our understanding of the interplay between the gut microbiota and CVD, emphasizing the role of cardiovascular aging and sarcopenia in HF. We examine the emerging evidence for the influence of the gut microbiome-derived metabolites in the development and progression of sarcopenia and HF. We also discuss several notable signal molecules and signaling pathways involved in both sarcopenia and HF and the contribution of gut microbiome during these processes.
MUSCLE FUNCTIONAL DECLINE AND WASTING IN HEART FAILURE
In patients with HF, muscle wasting (or “sarcopenia”) is one of the major causes of exercise intolerance, ventilatory inefficiency, chronotropic incompetence, and insulin resistance[5,7-11]. These conditions may induce other clinical disorders and eventually affect individuals’ quality of life. Although this phenomenon has been well recognized by clinicians over centuries, formal definitions of sarcopenia continue to evolve and are not standardized. Nevertheless, the contemporary focus on muscle wasting as a therapeutic target in HF began almost three decades ago when Coats et al. proposed the “muscle hypothesis” that cardiac impairment in HF may promote catabolism and reduce anabolic factors that can lead to skeletal muscle abnormality[15]. This hypothesis provided the explanation that HF patients develop progressive exercise intolerance above and beyond impairment in their cardiac reserve and may explain the increased sympathetic activation, vasoconstriction, endothelial dysfunction, and ultimately worsening of left ventricular contractile function. Based on this hypothesis, the imbalance of catabolic and anabolic processes in skeletal muscle metabolism is a key trigger of muscle wasting. Differential expression of the signaling molecules and pathways controlling muscle growth, structure, and myocyte homeostasis have been described in HF and the elderly. This review focus on skeletal muscle atrophy/wasting in aging and HF. Cardiac muscle atrophy, which is usually induced by cancer treatment, will not be discussed here. We would like to direct readers to these review articles for further reading[16,17].
Signal molecules involved in skeletal muscle metabolism
The signaling molecules that have been investigated include androgen, growth hormone (GH), insulin-like growth factor-1 (IGF-1), ghrelin, and myostatin, among many others [Figure 1]. Androgen, especially testosterone, is an anabolic hormone is naturally deceased with age in men. Low plasma testosterone level is associated with many metabolic syndrome and CVD[18] and is correlated with exercise intolerance in men with HF[19]. Although the impact of low plasma testosterone levels on skeletal muscle in older adult remains controversial, administration of testosterone has been observed to enhance physical performance of HF patients, e.g., quadriceps strength, walking test and oxygen consumption in several clinical studies[20-24]. Studies have demonstrated that testosterone can activate multiple cell signaling pathways associated with cardiac and skeletal hypertrophy[25,26]. Testosterone elicits its function via interacting with intracellular androgen receptor (AR). The activation of AR then exert its transcriptional function to active genes associated hypertrophy or interact with the signaling molecules involved in mammalian target of rapamycin (mTOR) and mitogen-activated protein kinases (MAPK) pathways[26,27], which are two important signaling transduction pathway in regulating muscle cell growth[28]. Insulin and IGF-1, are two well-known protein ligands that promote protein synthesis. They do so by activating the phosphoinositide 3-kinase-serine/threonine-protein kinase/mTOR (PI3K-AKT-mTOR) and MAPK pathways[28]. Activation of these pathways mediate the expression of myogenesis regulatory factors and contribute to dynamic protein balance in muscle cells[29-31]. Moreover, Activation of insulin/IGF-1 signaling pathways can ameliorate ubiquitin-proteasome-mediated proteiolysis as well as myostatin (MSTN) associated atrophic signaling that we will discuss below[30,32]. Ghrelin, an acylated peptide, stimulates the secretion of GH, cortisol, aldosterone, catecholamines, and prolactin. The release of GH can induce the secretion of IGF-1. It regulates appetite and food intake. Therefore, the reduction of the ghrelin level is linked to patients with HF and sarcopenia.
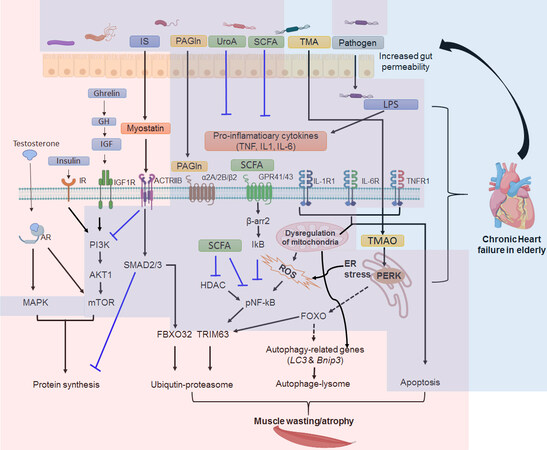
Figure 1. Gut microbiota dysbiosis-associated signaling pathways in muscle wasting and heart failure. Signaling pathways involved in skeletal muscle protein synthesis include androgen/testosterone, insulin and growth factors related pathways. Testosterone binds to its androgen receptor (AR) then together enter to nucleus to activate its downstream genes associated with muscle protein synthesis. It can also directly or indirectly influence mitogen-activated protein kinases (MAPK) and mammalian target of rapamycin (mTOR) pathways to control protein synthesis. IGF-1 and insulin elicit their protein sythesis function by binding to their receptors on cell membrane. Signaling pathways invovled in skeletal muscle wasting/atrophy include myostein-mediated transformation growth factor β signaling pathway, inflammation-assocaited signaling pathways. The ceullar mechamis asscoaited with skeletal muscle wasting/atrophy include the UPS proteiolsys pathway, autophage-lysome (ALP) and apopotsis. Dysregulation of mitochondria can trigger UPS, ALP and apoptosis leading muscle wasting/astrophy. Some gut microbial metabolites (GMM) and pathogens have anti-inflammation effect, such Short-chain fatty acids (SCFAs) and Urolithin A (UroA) and thus prevent muscle degredation. A variety of GMM and pathogens can trigger myostatin-mediated SMAD2/3 signaling pathway and inflammatory signaling pathways. One potential GMM that causes muscle wasting in the elderly have not been evulatued is TMAO. Background color in pink indicates cellular events and signaling pathways associated with muscle wasting, and background color in blue indicates those related to chronic heart failure. Please see the text for detail description. Please note that several signaling pathways such as MAPK, mTOR and SMAD2/3 are also involved in heart failure. However, the ligands trigger these signaling pathways leading to heart failure are different from those activate the pathways in muscle wasting. (Created with BioRender).
On the other hand, MSTN (also known as GDF8), belongs to the transformation growth factor β family functions via binding to the activin receptor type 2B recptor to activate actviate the mothers against decapentaplegic homolog 2 and 3 (SMAD2/3) signaling pathway and is a negative regulator of skeletal muscle growth. Overexpression of MSTN signaling in mice leads to muscle atrophy[33], whereas MSTN-knockout mice develop abnormal skeletal muscle hypertrophy[34]. It was reported that the MSTN level is increased in the hearts and in plasma of patients with HF[35,36] or in the skeletal muscle of rodent models with chronic HF[37]. Therefore, MSTN has been proposed as a potential therapeutic target in managing sarcopenia and HF.
Mechanisms involved in skeletal muscle metabolism
Besides pathways that link to skeletal muscle growth and development, studies have also supported the notion that increasing protein degradation or decreasing protein synthesis and cell death are major cellular events accruing in sarcopenia [Figure 1]. Specifically, three main cellular processes associated with skeletal muscle wasting, namely the ubiquitin-protease system (UPS), the autophagy-lysosome pathway (ALP), and apoptosis, are clearly altered in sarcopenic patients with HF.
Ubiquitin-protease system
Ubiquitin-protease system (UPS) is a major cellular proteolytic system responsible for degrading the misfolding proteins, cell cycle regulators, and transcription factors[38]. In UPS, the most well-known ubiquitin enzymes associated with muscle atrophy are two E3 ubiquitin-protein ligases, TRIM63 (Tripartite Motif Containing 63, or MuRF1), and FBXO32 (F-box only protein 32, or atrogin 1). The substrates for TRIM63 are myofibrillar proteins, such as titin, myosin heavy chain and light chain, and myosin-binding protein C, whereas FBXO32 targets factors that regulate proteins synthesis. Once recognizing their protein targets, they add multiple ubiquitin to the target proteins. The polyubiquitinated protein targets are then degraded via proteasomes[39]. Expression of TRIM63 and FBXO32 can be regulated by NF-kB (transcription factor nuclear factor-kB) and the FOXO (forkhead box group O) transcription factor family, which can be induced by pro-inflammation cytokines such as tissue necrosis factor alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1β (IL-1β). Interestingly, UPS and the pro-inflammation cytokine are also increased in the skeletal muscle of patients with HF, suggesting the common regulatory networks between HF and sarcopenia.
Autophagy-lysosome pathway
Another important system for protein breakdown in skeletal muscles is the ALP, which is a non-specific cellular process to degrade cytoplasmic damaged organelles and cellular molecules in the lysosome. Misregulation of autophagy in muscle causes mitochondrial damage, endoplasmic reticulum stress, impaired sarcomeric-protein turnover, and cell death[40]. It was reported that the system contributes to the protein degradation that occurs in denervated muscle in rodent models[41,42]. Physiologically, ALP is essential for muscle maintenance. Using fluorescence-activated cell sorting technique and naturally aged mice that contained green fluorescent protein on an autophagosome marker, LC3, García-Prat et al. demonstrated that the autophagy-lysosomal pathway prevents muscle stem cells from entering cell senescence[43] , which is a common cellular event present in the sarcopenic muscle at geriatric age[44]. Therefore, ALP may promote the stemness of stem cells allowing muscle regeneration efficiently. The function of ALP declines during aging in skeleton muscle, and the regeneration ability is also impaired in aging muscle. These studies demonstrated that ALP plays a critical role in maintaining muscle homeostasis and regeneration. However, several genetic models and human studies have shown the activation of ALP in muscle cells contributes to muscle wasting and atrophy. In addition, cathepsin L, a lysosomal protease in ALP system, was shown to be increased in catabolic muscle atrophy in rat models[45]. Constitutively active a transcription factor FOXO3, which leads to muscle atrophy, induces autophagy-related genes e.g., LC3 and Bnip3[46]. Using an oxidative stressed induced-muscle atrophy transgene mouse model, superoxide dismutase protein (SOD1G93A), Dobrowolny et al., showed that ALP was activated in SOD1G93A transgenic mice and reduced autophagy using shNRA-mediated knock-down LC3 restored muscle mass in transgene[47]. LC3 is also upregulated in different muscle atrophy states, such as cachexia, diabetes, and fasting[48]. Autophagy can also remove organelles selectively via a specific pathway, such as mitochondria via mitophagy, which is a necessary pathway to eliminate dysfunctional mitochondria and promote the renewal of these organelles. Dysregulation of mitochondria functions is well known for causing aging-associated diseases, including sarcopenia. In conclusion, the proper function of ALP is not only required for skeletal muscle homeostasis but also plays a role in muscle atrophy under catabolic conditions.
Myocyte apoptosis
Another mechanism associated with muscle atrophy and sarcopenia is myocyte apoptosis. Studies have been shown that a higher frequency of apoptosis of skeletal myocytes can be found in patients with HF or in animal models compared to the control groups[49-52]. Several common alterations of signaling pathways that occur in HF and sarcopenia can lead to cell apoptosis, particularly in the presence of the pro-inflammation signaling pathway. Evidence has been reported that the circulation level of TNF-α, C-reactive protein (CRP), and IL-6 correlated with a functional decline of skeletal muscle and muscle mass, suggesting a direct role of these molecules in sarcopenia[53,54]. Additionally, circulating concentrations of TNF-α trigger myocyte apoptosis in HF[55]. The lower grade of inflammation creates an oxidative stress environment that produces reactive oxygen and species (ROS), triggering the muscle atrophy program by activating the FOXO transcription factor family, which were discussed earlier. Therefore ROS has been linked to exercise intolerance, impaired mitochondria function[56]. Several studies have revealed that many mechanisms may contribute to the alteration of muscle metabolism in patients with HF. It was shown that oxidative phosphorylation is attenuated in patients with HF, energy transfer via mitochondrial creatine kinase is impaired, and overall ATP levels are reduced[57]. All these phenomena observed in patients with HF and sarcopenia may be linked to mitochondrial dysfunction in myocytes. Thus, understanding mitochondrial function and regulation may help us to better understand the mechanism underlying muscle wasting in sarcopenia and HF, which we will discuss further below.
CONTRIBUTIONS OF THE GUT MICROBIOME IN HEART FAILURE AND SARCOPENIA
The gut hypothesis of human disease can be traced back to nearly 2500 years ago when the ancient Greek physician Hippocrates claimed that “All disease begins in the gut” Increasing evidence supports the involvement of the gastrointestinal system in the pathogenesis of many human diseases, including HF and various aging-related conditions. First, alterations made in diet as well as aging itself can influence the composition of the gut microbiota. Second, the composition of the gut bacteria population is different between healthy and CVD subjects. Despite some well-recognized limitations, studies comparing the gut microbiota profiles in stool samples between healthy subjects and patients with CVD revealed the decreased diversity of the gut microbiota and alteration of microbiota composition[58]. Lastly, the gut metabolite generated by the gut microbiome can affect the host’s physiology and pathophysiology. For example, lipopolysaccharide (LPS) and trimethylamine N-oxide (TMAO), can induce inflammation while short-chain fatty acids (SCFAs) and bile acids (BA) can effect on host metabolism[59].
The gut-heart axis: interplay between the gut microbiome and cardiac aging and disease
Various studies over the past decade have linked gut microbiota with pathogenesis of heart disease, especially in the setting of HF[60-62]. Alterations of the gut microbiota can lead to the development of risk factors for atherosclerotic vascular disease and directly impact pathogenic disease progress such as acute coronary syndromes and HF[63]. In the gut-heart axis scenario, gut bacteria and their endotoxin products, i.e., LPS, may translocate to the host circulation due to the impairment of the gut barrier function that abnormally increased the intestinal permeability, which occurs in patients with HF. This bacterial translocation may trigger low-grade systemic inflammation, alter the immune system, and can lead to increased disease severity in patients with HF. Several animal studies and association studies in humans support this hypothesis. By comparing biomarkers profiles for patients who had chronic HF with or without recent-onset peripheral edema and healthy individuals, Niebauer et al. reported that the endotoxin concentration and pro-inflammatory cytokine are significantly increased in patients who had chronic HF with peripheral edema but not in other groups[64]. The increased endotoxin can be normalized by a short-term diuretic treatment but not for cytokine[64]. Recently, Beale et al. demonstrated that patients with HF with preserved ejection fraction had a depletion of SCFA-producing bacteria that generate SCFA, known to have a beneficial and vital role for cardiovascular homeostasis in small cohorts from two independent communities[62]. Several microbiome-derived metabolites have been demonstrated their effects in the heart. We will discuss them below.
Trimethylamine N-oxide
Among the gut microbiome-derived metabolites in human diseases, TMAO is the most notable one. TMAO is the compound generated from trimethylamine (TMA)-containing dietary nutrients such as choline, betaine, and L-carnitine. It was recently demonstrated that a microbial gene cluster, gbu, is responsible for converting an intermediate product of dietary carnitine, γ-butyrobetaine to TMA[65]. The circulating TMA is then converted into TMAO by FMO3 (hepatic flavin monooxygenase 3) in the hosts’ liver. Work from our groups and others has demonstrated that the TMA-TMAO pathway as a novel connection between diet the gut microbiota and the risks of atherosclerosis[66,67] and thrombosis[68-70]. For example, elevation TMAO level in plasma was found to be associated with an increased risk of occurrence major adverse cardiovascular events (MACEs) in a large cohort study[60]. A smaller cohort study also revealed that the increased plasma level of TMAO was associated with acute coronary syndromes and can be used as a prognostic marker for MACEs[71]. Additionally, using an untargeted metabolomics approach, our group has identified that TMAO and N6,N6, N6-trimethyl-L-lysine (trimethyl lysine, TML), a TMAO-producing nutrient precursor, can serve as predictors for MACEs[67,72]. Moreover, association studies linked TMAO with an increasing level of CRP and LPS to contribute to chronic inflammation and endothelial dysfunction[68,73].
How does TMAO cause cardio-metabolic diseases? Chen et al., demonstrated that the pathophysiological level of TMAO can promote ER stress by directly binding to the endoplasmic reticulum stress kinase PERK (EIF2AK3) and inducing the expression level of FOXO1 in hepatocytes to cause hyperglycemia and metabolic dysfunction[74]. Other potential mechanisms for TMAO have been discussed, such as activation of pro-inflammation pathway causing fibrosis[53], promoting microvascular dysfunction in the heart tissues of patients with HF without the presence of coronary disease, and having a synergized effect on neurohormonal derangements[75]. However, mechanic studies are needed to further demonstrate the direct detrimental effect of TMAO in these processes in humans. Meanwhile, TMAO has been associated to cardiovascular aging. Plasma level of TMAO was shown to be increased in the elderly, both in humans and rodent models[76]. The elevated circulating TMAO level was shown to contribute to endothelial dysfunction, a characteristic of the aging process, in aged rats, possibly through increasing vascular inflammation and oxidative stress[76]. Long-term feeding mice with TMAO induces aging-like vascular endothelial dysfunction through reduced nitro oxide (NO) bioavailability and promoted oxidative stress in young mice and healthy humans[77]. The studies suggested that TMAO may play a role in the development of age-related diseases.
Short-chain fatty acids
According to the American Heart Association diet and lifestyle recommendations that high-fiber diets have a beneficial association with a lower risk of developing CVD[78]. Although the beneficial effects of high-fiber diets remain to be explored, recent attention has been focused on the effect of SCFAs on the host’s physiology. As mentioned above, SCFAs are the end-products from the fermentation processes of gut microbiota and are derived from the non-digestible soluble fibers such as cellulose and other complex carbohydrates that can’t be used by the host[79]. High-fiber diets increase the diversity of the gut microbiome and are favored for SCFA-producing bacteria, while low-fiber diets are linked to gut dysbiosis and hypertension[80]. The majority of SCFA products in the gut are acetate, propionate, and butyrate. It is now clear that SCFAs have anti-inflammatory effects as well as profoundly regulate the host’s immune system. How does SCFA modulate inflammation and the host’s immune system? Strong evidence support that SCFAs act as signaling molecules binding to their well-known G-protein-coupled receptors (GPCR), GPR41, GPR43[81], GPR109A[82] and OLFR78[83] in animal models. However, the precise mechanism remains unclear. Mechanically, it was demonstrated that SCFAs bind to their GPCR, e.g., GPR41/43, to regulate its downstream signal molecules such as activation of p38 and suppression of AKT and ERK pathways. As a result, it downregulates the expression of inflammatory cytokines such as IL-1α, IL-1β and Intercellular Adhesion Molecule 1, reduces the production of chemokine, and attenuates TNF-α associated inflammation pathways[84,85]. Moreover, the activation of GPR41/43 can increase the interaction of β-arrestin with I-kBα. This interaction, thus, reduces the phosphorylation of NF-kB and downregulation of IL-1β and Monocyte chemoattractant protein-1 secretion[86,87]. The beneficial cardiac effects of SFCAs were also documented. A study done by Marques et al. demonstrated that dietary fiber and acetate supplementation in deoxycorticosterone acetate (DOCA)-salt hypertension mouse model not only increases SCFA-producing bacteria in the gut but also reduces blood pressure, cardiac hypertrophy and fibrosis, and improves cardiovascular function[88]. Similar results were also reported in a cardiac injury rat model induced by angiotensin II (Ang II). Sodium butyrate (NaBu) feeding in an Ang II-induced cardiac injury rat model attenuated cardiac hypertrophy and suppressed fibrosis and inflammation. Interestingly, this inhibition of inflammation by NaBu may work through the cyclooxygenase (COX)/prostaglandin E2 (PEG2) pathway, which is associated with the ERK pathway[89]. The anti-inflammation effect of SCFAs may also act at the epigenetic level. It is known that butyrate is a potent histone deacetylase (HDAC) inhibitor. The inhibition of HDAC increases acetylation of histones that enhance the accessibility of the chromatin and thus upregulation of gene expression[90]. In vitro cell culture experiment showed that Nabu suppressed Ang II-induced cardiac hypertrophy by inhibiting COX/PGE2 pathway in an HDAC5/HDAC6-dependent manner[62]. SCFA treatment in LPS or TNF-α exposed human umbilical vein endothelial cells reduced the inflammation-associated genes, such as IL-6 and vascular cell adhesion molecule 1 as well as the HDAC activity[91]. It was demonstrated that butyrate elicits its anti-inflammatory effects via the inhibition of HDAC in a bone marrow-derived macrophages (BMDM) model stimulated by LPS. The recruitment of polymerase II to the promoter of the inflammation genes, e.g., IL-6, interleukin-12, and Nos2, was reduced compared to the non-butyrate treated LPS-BMDMs[92]. Taken together, SCFAs act in the host to modulate its metabolism and immune function and contribute to the host’s health. SCFA supplementation can improve metabolic status in HF conditions. However, more clinical studies are needed to support such intervention in humans.
Bile acids
Bile acid (BA), especially the secondary BA converted by the gut microbiota, is linked to HF. The primary BA, which is normally stored in the gallbladder, is synthesized in the liver from cholesterol and forms the major component of bile. The function of primary BA is to emulsify dietary fats and to facilitate the absorption of lipids and lipid-soluble vitamins. Because diet and low-density lipoprotein (LDL)-cholesterol (LDL-C) are risk factors for developing CVD, dietary strategies to reduce LDL-C levels to prevent disease are a recommendation for people with a high level of blood lipids hyperlipidemia and hypertriglyceridemia. The impact of diet and lipid metabolism of in CVD has been reviewed[93,94]. In this review, we focused on the gut microbiota-derived BA on CVD and aging. The secondary BAs is included deoxycholic acid (DCA), lithocholic acid (LCA), hyodeoxycholic acid, and ursodeoxycholic acid are converted by gut microbiota-derived enzymes in the colon[95]. Bile salt hydrolase and 7α and 7β dehydoxylase are responsible for the generation of secondary BAs in the gut[96].
It is well known that secondary BAs act as signaling molecules that interact with cell-surface and nuclear hormone receptors to modulate dietary lipid metabolism[97] and energy homeostasis[98] and potentially affecting CVD pathogenesis. Elevated serum BAs have been long speculated to affect cardiac function. Several in vitro cell culture studies showed that the cardiomyocyte function, such as contraction action potential, was impaired upon various BAs treatment[99-103]. Additionally, in a BA-overload mouse model in which Fxr (farnesoid X receptor) and Shp (small heterodimer partner), two critical nuclear receptors responsible for maintaining bile acid homeostasis, are deleted, Desai et al. showed that the double knockout mutants (DKO) display cardiac hypertrophy, bradycardia and exercise intolerance[104]. When treating this DKO mouse with cholestyramine, a bile acid-binding resin known to reduce the circulating BA pool, the cardiac dysfunction phenotypes were restored, suggesting BA is directly responsible for the cardiac dysfunction presented in DKO mice[104]. A small clinic exploratory study reported that the ratio of secondary to primary BAs was increased in patients with chronic HF[105]. The ratio was associated with reducing overall survival on univariate analysis but not significant after adjustment for clinical characteristics and NT-proBNP. Whether changes in bile acids promote disease progression and sarcopenia in HF and aging beyond their metabolic effects remains unknown at this point. Clearly, more clinical studies with a large sample size are required to confirm the data as well as explore the regulation and potential role of BA metabolism in HF.
Dietary polyphenols
Dietary polyphenols, particularly ellagitannins (ETs), have been shown to have an antioxidant and anti-inflammatory effect that may have a beneficial role on the skeletal muscle and heart during the aging and disease process[106,107]. ETs can be found in pomegranates, berries, walnuts, and oak-aged red wines[108]. Urolithin A (UroA) and urolithin B (UroB) are two major gut metabolites produced from ETs-containing foods[109]. Administration of Uro A and UroB was shown to suppress the pro-inflammatory cytokine fractalkine and a sarco/endoplasmic reticulum Ca2+ ATPase, SERCA2, in the mouse heart as well as restore the mechanical properties and calcium dynamics of cardiomyocyte[110]. In a surgical induced myocardial reperfusion injury mouse model, it was demonstrated that UroA reduced ROS reaction via controlling PI3/AKT pathway and enhancing antioxidant activity[111]. Additionally, Ghosh et al. showed that UroA supplementation delayed the onset of muscular aging by increasing adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide (NAD+) levels in murine skeletal muscle[112]. It was shown that UroA elicits its beneficial effect via enhancing mitochondria health, particularly via increasing mitochondria mitophagy. However, the mechanical insight of how UroA improves mitochondria health in cardiac aging, heart disease and sarcopenia remains to be explored.
Phenylacetylglutamine
Other gut microbial metabolites (GMM) associated with CVD have recently been reported. Using untargeted metabolomics, Nemet et al. reported that the plasma metabolite Phenylacetylglutamine (PAGln) was associated with CVD and major adverse cardiac events, which confirmed prior observations in patients with chronic kidney diseases where PAGln was considered a uremic toxin[113]. The microbial gene, porA, is responsible for covert dietary phenylalanine to PAGln. It was shown that PAGln accelerated platelet clot formation and enhanced thrombosis potential in mice, likely via through its homology to adrenergic receptor ligands to the α2A, α2B, and β2 adrenergic receptors[113]. Mechanistically, how PAGln can drive disease progression in CVD remains an area of active investigation.
Other protein-bound uremic toxin
Two of most investigated protein-bound uremic toxin (PBUTs), indoxyl sulfate (IS) and p-cresyl sulfate (pCS), have been shown to predict clinic outcomes in patients with chronic kidney and heart diseases and correlate with cardiovascular events and all-cause mortality[114-116]. IS is derived from bacterial metabolism of dietary tryptophan to indole and pCS is synthesized by intestinal bacteria from tyrosine and phenylalanine. Both substances are normally cleared by renal proximal tubular secretion but accumulate in the circulation once renal function is impaired[117]. Both substances also have been demonstrated to have cardiac adverse effect. The profibrotic prohypertrophic and pro-inflammatory effect of IS was first demonstrated by Krum group more than decade ago in vitro cell culture system. Treatment of cardiac cells stimulate cardiac fibroblast collagen synthesis, hypertrophy and activate the gene expression level for key inflammatory cytokine such as TNF-α, IL-6, and IL-1β[118]. Animal study using Dahl salt-resistant normotensive IS-administered rats also demonstrated that IS aggravates cardiac fibrosis and hypertrophy and enhance the oxidative stress[119]. The parent from of PCS, p-cresol, was shown to activate of Ca2+-dependent protein kinase Cα that lead to reduction of the contraction rates and disassembling of connexin 43 gap in the culture cardiomyocytes[120]. Moreover, it was shown that PCS promote cardiac apoptosis, affected function of the left ventricle of the heart in the mice that underwent 5/6 nephrectomy[121]. PBUTs play a role in vascular dysfunction[122]. However, their effect on cardiac aging have not been evaluated. Because PBUTs also promote cardiac inflammation or induce oxidative process, which are two major cellular events occurred in aging process, one would assume these compounds may play a role in cardiac aging. Interestingly, it was recently reported that Klotho, an anti-aging protein with reno/cardio-protective effects, can suppress the IS-induced inflammatory response to protect against renal fibrosis and cardiac hypertrophy by promoting M2 macrophage polarization[123]. Interestingly, Klotho deficiency was recently shown to cause cardiac aging via affecting the molecules involved in the against oxidative stress[124]. The restoring Kloth function might be an alternative intervention for organ dysfunction assorted with aging.
The gut-muscle axis: interplay between gut microbial metabolism and muscle wasting
Emerging evidence in animal models has shown that gut microbiota-derived metabolites have a direct effect on muscle wasting. Siddharth et al., found that aged rat with sarcopenia displayed a distinct gut microbiome compared to rats with a normal muscle mass[125]. The muscle dysfunction is correlated with the downregulation of genes involved in the vitamin, carbohydrate, protein, and lipid biosynthesis and metabolism, which may contribute to the regulation of muscle mass. In an acute leukemia-induced muscle wasting mouse model (BAF3), feeding mice with Lactobacillus species, which were reduced in the BAF3 mice versus the control mice, reduced the inflammation and attenuated muscle atrophy[126]. In another study, treating mice with Faecalibacterium prausnitzii reduced adipose tissue inflammation, increased gastrocnemius muscle mass, and the expression of mitochondrial respiratory chain protein in high-fat-fed mice[127]. The administration of Lactobacillus reuteri to a cancer cachexia mouse model, which exhibited adipose tissue and muscle atrophy phenotypes, can increase muscle mass and fiber size[128]. Feeding naturally aged mice with butyrate, a gut microbiota-derived SCFAs and HDAC inhibitor, increased muscle fiber cross-sectional area and prevented intramuscular fat accumulation in the old mice[129]. We have discussed the effect of SCFA above. In a genetic knockout mouse model in which ghrelin, a hormonal peptide functions as a GH stimulator, was deleted, it was shown that the old ghrelin KO mice developed microbial dysbiosis and worsened muscle atrophy compared to the former wildtype mice under fast-induced conditions[130]. All these studies highlight the role of gut microbiota-derived metabolites in regulating skeletal muscle metabolism.
Inflammation signaling pathway is one of the key factors of the imbalance in skeletal muscle metabolism leading to muscle wasting. Therefore, GMM that can modulate inflammation signaling pathways, such as SCFAs, can affect normal skeletal muscle metabolism and cause muscle wasting. Indeed, various studies have been demonstrated that SCFAs play a profound role in regulating muscle mass and physical function[131]. There are studies that suggest that liver disease, which causes abnormal production of BAs, may lead to muscle wasting and cachexia[132,133]. However, currently, there is no study evaluating the GMM-BAs associated with sarcopenia. Although the role of TMAO in CVD and other metabolic diseases is studied extensively, the link between TMAO and muscle wasting in sarcopenia remains unclear. An association study recently reported that the plasma level of TMAO was significantly and independently increased in frail patients with CVD than in non-frail ones[134]. However, since the study did not collect other important data for evaluating the relationship between dietary and muscle wasting, such as nutritional data and body composition of patients, further studies with such information are required to study the role of TMAO in sarcopenia.
Interestingly, gut microbiome-derived uremic toxins, IS, were shown to be associated with sarcopenia[135,136]. By treating C2C12 myoblast cells with six-protein-bound uremic toxins, Enoki et al. identified that IS may act a potent uremic toxin that promotes developing skeletal muscle atrophy[135]. It was shown that IS inhibited myoblast proliferation as well as myotube differentiation and formation by inducing oxidative stress-mediated expression of myostatin and atrogin-1 and inflammatory cytokine expression IL-6 and TNF-α in muscle cells[135]. Moreover, chronic administration of IS increased myostatin and atrogin-1 expression levels and decreased muscle mass in mice. Similar results were also reported by another study, and it was shown that cell apoptosis was increased in IS-treated myoblasts[136]. Additionally, Sato et al. demonstrated that IS induced metabolic alteration in C2C12 myotubes by upregulating glycolysis, upregulated glycolysis, pentose phosphate pathway and glutathione metabolism and decreased energy generation pathways[137]. Studies suggest that IS may serve as a key pathogenic factor for sarcopenia. Understanding the mechanism by which the IS-induced muscle atrophy can open up the potential therapeutic strategies for treating muscle wasting and sarcopenia.
Several clinical studies also revealed the importance of GMM in muscle wasting. A recent study done by Picca et al. showed that old people with physical frailty and sarcopenia have a different abundance of the gut microbiome, suggesting the changes in the gut microbial composition is associated with sarcopenia[138]. In a randomized, double-blind trial study, in which the efficacy of prebiotic Darmocare Pre® was evaluated in sixty older individuals aged 65 and older. Although the prebiotic did not significantly improve frailty in order adults, the score for exhaustion and handgrip strength was improved[139]. However, in a three-month intake of synbiotic supplementation study, the body composition and inflammatory status did not change in the elderly but showed an improvement of hydration status. Additionally, Aoyagi et al. reported that drinking fermented milk with different frequencies does not alter muscle mass in the elderly[140]. It seems that there is a discrepancy among the clinical trial studies. However, all these human studies were performed on a variety of populations and conditions with different measurement criteria. Therefore, a large cohort multicenter study with systematic evaluation methods should be performed in the future to confirm the beneficial effect of probiotics on improving muscle function and physiology in sarcopenia.
GUT MICROBIAL METABOLITES AND MITOCHONDRIAL FUNCTION IN CARDIAC AGING AND MUSCLE WASTING
Both heart and skeletal muscle are high-energy demand organs. They use ATP as the energy for myocyte contraction to allow skeletal muscle movement and locomotion and the heart to pump the blood to the whole body. Each heartbeat costs about 300 mg of ATP in humans, while the demand of ATP increases more than 100 times in skeletal muscle during intensive exercise[141,142]. Mitochondria are double-membrane organelles and the energy production center of the cells. They also play a vital role in many cellular processes, including cellular redox homeostasis, ion homeostasis, calcium storage and homeostasis and regulation of cell death and survival. Mitochondria produce ATP via oxidative phosphorylation (OXPHOS), which takes place in the inner mitochondrial membrane. During OXPHOS, electrons derived from NADH and FADH2 combine with molecular oxygen by entering the electron transport chain. During this process, protons are pumped to the intermembrane space of mitochondria that produce a membrane potential serving as energy to the synthesis of ATP from ADP. OXPHOS also generate byproduct reactive oxygen species (ROS), which are known to regulate many physiological functions in normal and disease process. Mitochondria also serve as sensors and regulators of calcium signaling, which is an essential mechanism to maintain intracellular calcium homeostasis as well as regulate energy production[142,143].
Mitochondrial dysfunction has been considered as an underlying cause of myocyte loss during cardiac aging, failure, and sarcopenia[144,145]. Studies from animal and human studies link mitochondrial energetics and control of muscle mass and cardiac aging. The Baltimore Longitudinal Study of Aging demonstrated that mitochondrial function declines with age and may contribute to age-associated loss of muscle function and cardiorespiratory fitness[146]. Using unbiased transcriptomic and targeted quantitative metabolomics profiling, Lai et al., showed that mitochondrial energy metabolic derangements, such as the lower amount of many Kerbs cycle intermediates and elevation in lactate and acylcarnitine species, occur during the early development of HF induced by pressured overload mouse model, suggesting a bottleneck of carbon flux into mitochondria[147].
Mitochondria dynamics, which involves mitochondria fusion and fission, is important for mitochondrial maintenance. The imbalance of the fusion and fission process is a feature for cell senescence. Evidence from mouse genetic models in which genes encoded mitochondrial fusion or fission proteins were mutated also demonstrated the importance of mitochondria during aging and sarcopenia. For example, the ablation of OPA1 mitochondrial dynamin like GTPase induced FOXO signaling and unfolded protein response which caused catabolic program of muscle loss and systemic aging[148] and mice without mitochondrial pro-fusion factors such as Mfn2 (mitofusin 2) in skeletal muscle developed significant defects of muscle growth and sarcopenia[149]. The impact of mitochondrial dynamics in aging and sarcopenia was reviewed[150,151].
In aging or stressed myocytes, the elevation of ROS levels, increasing of oxidative stress and overload of Ca2+ uptake are present in mitochondria. The elevation of these cellular events causes the opening of mitochondrial permeability transition pore (mPTP) and thus loss of the mitochondrial membrane potential. The high ROS level and loss of the mitochondria membrane potential can cause reduction of ATP production, trigger mitophagy, cell death and impaired mitochondrial biogenesis. These cellular events can affect muscle metabolism leading to muscle atrophy, cardiac aging and failure. For example, excessive mitochondrial ROS trigger protein degradation systems, ALP and UPS[152,153] and reduced ATP production can activate the AMP kinase -FOXO3 pathways leading to upregulation of protein degradation systems[46,154] that were known for affecting muscle metabolism as we discussed earlier.
In dysfunctional mitochondria, the microconidia contents, such as mitochondria genomics, ATP, and transcription factors, may be released to the cytosol to trigger various pro-inflammatory signaling. These leaking microdontia contents serve as damage-associated molecular patterns (DAMP) to activate nucleotide-binding oligomerization domain (NOD)-like receptor protein 3 (NLRP3) associated inflammasomes intracellular multiprotein complexes and enhance the inflammatory response of the cells[155].
Alteration of mitochondrial biogenesis is another feature of age-related mitochondrial dysfunction. Mitochondrial biogenesis generates new mitochondria through multiple transcriptional regulatory pathways, the transcription of nuclear genes for biogenesis, import of nuclear-encoded mitochondria proteins and transcription and replication of mtDNA[156]. PGC-1α (peroxisome proliferator-activated receptor-gamma coactivator α) is the master regulator in mitochondrial biogenesis. PGC-1α activates the transcription and transcriptional activity of the nuclear respiratory factor-1 and -2 (Nrf-1 and Nrf-2) and cooperates with NRF1/2 and related receptor alpha (ERRα) to activate mitochondrial biogenesis cascade. NRFs then activate the mitochondrial transcription factor A (Tfam) to activate the mitochondrial genome by triggering replication and transcription of the corresponding regions of mtDNA[157]. Evidence from human and animal studies also revealed that mitochondria biogenesis plays a role in pathophysiologic processes in sarcopenia and cardiac aging. The aging heart displays a reduced level of PGC-1α along with its downstream targets involved in mitochondria biogenesis[158-160]. The decline of mitochondria biogenesis was observed in the aging skeletal muscle and heart. In a multi-ethnic study of human sarcopenia versus age-matched controls in muscle biopsies, the transcriptome profiles of genes involved in mitochondria biogenesis, e.g., PGC-1α and ERRα nuclear receptor, were reduced in sarcopenic individuals[161]. Another study by Kong et al. also observed the key biogenesis regulator, PGC-1α, and its downstream effector and a major mitochondrial deacetylase, SIRT3[162], were dramatically reduced in the muscle fibers taken from elderly participants compared to young participants[163]. Similar findings were also found in a spontaneous senescence-accelerated mouse (SAM) prone 8 (SAMP8) aging mouse model, whereas the expression of PGC-1α, Nrf-1 and Tfam were significantly decreased[164]. Taken together, proper regulation of mitochondrial function and physiology, including mitochondria dynamic, mitophagy and biogenesis, is important in healthy cardiac and skeletal muscle aging. Modulating the pathways associated with these processes may effectively improve the aging phenotype in the heart and skeletal muscle.
Studies on GMM and mitochondrial function in myocytes have been reported. It was shown that germ-free mice have a lower mtDNA content, reduced mitochondrial biogenesis markers, and oxidative phosphorylation complexes gene expression in skeletal myocytes, indicating alteration of mitochondria function and metabolism in skeletal muscle of germ-free mice[165]. Munukka et al. gave SCFA-producing bacteria Faecalibacterium prausnitzii to mice that were fed with a high-fat diet (HFD) and resulted in upregulation of the expression of mitochondrial respiratory chain complex and increased gastrocnemius muscle mass[127]. Administration of UroA in mice fed with HFD improves skeletal muscle function in aged mice compared to that in the control HFD-fed mice only[166]. However, treating mice with TMAO impaired b-oxidation in cardiac mitochondria, deceased OXPHOS, and pyruvate metabolism[167]. Cardiomyocytes treated with a pathological concentration of chenodeoxycholic acid, a bile acid, suppressed the expression of genes involved in fatty acid oxidation in mitochondria[104]. GMM-derived uremic toxin, IS, was shown to cause mitochondrial dysfunction and ATP shortage in muscle cells[137]. These preclinical studies demonstrated that GMM have a profound impact on mitochondrial function in skeletal muscle and hearts.
POTENTIAL THERAPEUTIC STRATEGIES FOR IMPROVING GUT DYSBIOSIS IN AGING POPULATION
It is clear that aging is linked to gut dysbiosis[168,169]. As discussed above, several GMMs can induce inflammation, which is associated with multiple diseases in the elderly. Chronic low level of inflammation is a hallmark of human aging. Therefore, strategies that can reduce the production of GMM-associated inflammation or promote bacteria that can attenuate inflammation may hold promising ways to improve the overall physiology performance of old adults. Here we list two potential gut microbiome-based therapeutic strategies for the aging population and discuss their limitations.
Physical exercise strategy
Aging is associated with reduced muscle function and atrophy. The correlation between physical exercise and improvement of muscle size and overall physical function is well established. Evidence also supports the notion that physical exercise is beneficial for improving aerobic capacity, metabolic regulation cardiovascular function, and muscle mass and function[170-173]. Additionally, several studies have shown that there is a strong association between exercise and a health-beneficial gut microbiome, suggesting physical exercise increase the growth of beneficial gut microbe[174-177]. However, such relationships remain associative in nature and more human studies are required to confirm if the beneficial effects of physical exercise on physical performance and cardiac function are modulated by gut microbial metabolism.
Nutritional strategy
Because the production of GMM is highly related to what we eat daily and some GMM is associated with CVD and aging-associated diseases, a nutrition-based approach maybe one of the most efficient ways to modify the gut microbiome and GMM. It is well established that high-fiber diets favor SCFA-producing gut microbe, and may promote an anti-inflammation effect (see Section The gut-heart axis: interplay between the gut microbiome and cardiac aging and disease). Thus, high-fiber diets are becoming an important strategy to improve health conditions in our daily life, particularly for the elderly. While several animal studies have demonstrated a high-fiber diet can increase muscle fiber and improve physical function, it remains unclear if the beneficial effects of high-fiber diets on increasing muscle mass and function are directly mediated by gut microbial metabolism.
Another nutritional strategy is calorie restriction (CR). CR is a primary dietary intervention shown to lower the risk of CVD and increase the healthy life span[178]. Animal studies indicate that CR can promote the growth of several health-beneficial microbes[179-182]. For example, Fraumene et al. have demonstrated that CR fosters the growth of Lactobacillus in a rat study[183]. Although animal and human trial studies suggest that CR may be a feasible strategy to improve the physical performance of older adults, more research remains needed to determine the suitably restricted calories for each older individual to avoid deteriorating their physical condition.
Lastly, oral intake of probiotics, prebiotics, or synbiotics have been considered as an effective and non-invasive option for countering aging pathways. Theoretically, this strategy can increase the populations of health-beneficial gut microbes such as bifidobacteria or lactobacilli in older adults[184-186]. However, the positive effect is more significant on mental and digesting system health[185-188]. For example, Inoue et al., conducted a randomize, double-blind placebo control trial by providing Probiotic bifidobacteria supplementation or placebo to 38 elderly for 12 weeks, and they found that probiotics supplement combined with moderate resistance exercise may improve the mental condition and bowel movement[188]. In contrast, the protective effect against sarcopenia and HF from consuming probiotics, sytiotics or prebiotics remains inconclusive, and the presence of an intact gut microbiome may deter any substantial changes in gut microbial populations.
CONCLUSION
The gut-muscle and gut-heart axis play key roles pathophysiology of aging-associated heart disease and sarcopenia. While there is ample evidence showing the correlation between GMM and aging-associated heart and muscle diseases, precisely how they interact to promote sarcopenia remains poorly understood. More mechanical and in vivo studies are necessary to dissect the underlying mechanisms of how GMM regulates myocyte (in the heart and skeletal muscle) during aging and pathophysiological processes at cellular and molecular levels. Nevertheless, there is tremendous promise for dietary or therapeutic interventions to target gut microbiome-associated metabolic pathways with GMM as mechanism biomarkers that can identify underlying pathophysiologic processes that link sarcopenia with HF and aging.
DECLARATIONS
Authors’ contributionsWrote the manuscript: Liu CF, Tang WHW
Made all figures: Liu CF
Availability of data and materialsNot applicable.
Financial support and sponsorshipDr. Tang was supported by grants from the National Institutes of Health (R01HL126827).
Conflicts of interestDr. Liu has no relationships to disclose. Dr. Tang is a consultant for Sequana Medical A.V., Cardiol Therapeutics Inc, Genomics plc, Zehna Therapeutics Inc, and has received honorarium from Springer Nature for authorship/editorship and American Board of Internal Medicine for exam writing committee participation.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Virani SS, Alonso A, Aparicio HJ, et al. Heart disease and stroke statistics-2021 update: a report from the american heart association. Circulation 2021;143:e254-743.
2. Tsao CW, Aday AW, Almarzooq ZI, et al. Heart disease and stroke statistics-2022 update: a report from the american heart association. Circulation 2022;145:e153-639.
3. Jousilahti P, Vartiainen E, Tuomilehto J, Puska P. Sex, age, cardiovascular risk factors, and coronary heart disease: a prospective follow-up study of 14 786 middle-aged men and women in Finland. Circulation 1999;99:1165-72.
7. Cruz-Jentoft AJ. Sarcopenia: revised European consensus on definition and diagnosis. Age Ageing 2019;48:16-31.
8. Bekfani T, Pellicori P, Morris DA, et al. Sarcopenia in patients with heart failure with preserved ejection fraction: Impact on muscle strength, exercise capacity and quality of life. Int J Cardiol 2016;222:41-6.
9. Emami A, Saitoh M, Valentova M, et al. Comparison of sarcopenia and cachexia in men with chronic heart failure: results from the Studies Investigating Co-morbidities Aggravating Heart Failure (SICA-HF). Eur J Heart Fail 2018;20:1580-7.
10. Streng KW, Nauta JF, Hillege HL, et al. Non-cardiac comorbidities in heart failure with reduced, mid-range and preserved ejection fraction. Int J Cardiol 2018;271:132-9.
11. Tucker WJ, Haykowsky MJ, Seo Y, Stehling E, Forman DE. Impaired exercise tolerance in heart failure: role of skeletal muscle morphology and function. Curr Heart Fail Rep 2018;15:323-31.
13. Haehling S, Ebner N, Dos Santos MR, Springer J, Anker SD. Muscle wasting and cachexia in heart failure: mechanisms and therapies. Nat Rev Cardiol 2017;14:323-41.
14. Biagi E, Franceschi C, Rampelli S, et al. Gut microbiota and extreme longevity. Curr Biol 2016;26:1480-5.
15. Coats AJ, Clark AL, Piepoli M, Volterrani M, Poole-Wilson PA. Symptoms and quality of life in heart failure: the muscle hypothesis. Br Heart J 1994;72:S36-9.
16. Murphy KT. The pathogenesis and treatment of cardiac atrophy in cancer cachexia. Am J Physiol Heart Circ Physiol 2016;310:H466-77.
17. Sweeney M, Yiu A, Lyon AR. Cardiac atrophy and heart failure in cancer. Cardiac Failure Rev 2017;03:62.
18. Brand JS, Rovers MM, Yeap BB, et al. Testosterone, sex hormone-binding globulin and the metabolic syndrome in men: an individual participant data meta-analysis of observational studies. PLoS One 2014;9:e100409.
19. Jankowska EA, Filippatos G, Ponikowska B, et al. Reduction in circulating testosterone relates to exercise capacity in men with chronic heart failure. J Card Fail 2009;15:442-50.
20. Pugh P. Acute haemodynamic effects of testosterone in men with chronic heart failure. Eur Heart J 2003;24:909-15.
21. Pugh PJ, Jones RD, West JN, Jones TH, Channer KS. Testosterone treatment for men with chronic heart failure. Heart 2004;90:446-7.
22. Malkin CJ, Pugh PJ, West JN, van Beek EJ, Jones TH, Channer KS. Testosterone therapy in men with moderate severity heart failure: a double-blind randomized placebo controlled trial. Eur Heart J 2006;27:57-64.
23. Caminiti G, Volterrani M, Iellamo F, et al. Effect of long-acting testosterone treatment on functional exercise capacity, skeletal muscle performance, insulin resistance, and baroreflex sensitivity in elderly patients with chronic heart failure a double-blind, placebo-controlled, randomized study. J Am Coll Cardiol 2009;54:919-27.
24. Iellamo F, Volterrani M, Caminiti G, et al. Testosterone therapy in women with chronic heart failure: a pilot double-blind, randomized, placebo-controlled study. J Am Coll Cardiol 2010;56:1310-6.
25. Wilson C, Contreras-Ferrat A, Venegas N, et al. Testosterone increases GLUT4-dependent glucose uptake in cardiomyocytes. J Cell Physiol 2013;228:2399-407.
26. Wu Y, Bauman WA, Blitzer RD, Cardozo C. Testosterone-induced hypertrophy of L6 myoblasts is dependent upon Erk and mTOR. Biochem Biophys Res Commun 2010;400:679-83.
27. Basualto-Alarcón C, Jorquera G, Altamirano F, Jaimovich E, Estrada M. Testosterone signals through mTOR and androgen receptor to induce muscle hypertrophy. Med Sci Sports Exerc 2013;45:1712-20.
28. Egerman MA, Glass DJ. Signaling pathways controlling skeletal muscle mass. Crit Rev Biochem Mol Biol 2014;49:59-68.
29. Rommel C, Bodine SC, Clarke BA, et al. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol 2001;3:1009-13.
30. Sacheck JM, Ohtsuka A, McLary SC, Goldberg AL. IGF-I stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1. Am J Physiol Endocrinol Metab 2004;287:E591-601.
31. Haddad F, Adams GR. Inhibition of MAP/ERK kinase prevents IGF-I-induced hypertrophy in rat muscles. J Appl Physiol (1985) 2004;96:203-10.
32. Stitt TN, Drujan D, Clarke BA, et al. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Molecular Cell 2004;14:395-403.
33. Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA 2001;98:9306-11.
34. McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 1997;387:83-90.
35. George I, Bish LT, Kamalakkannan G, et al. Myostatin activation in patients with advanced heart failure and after mechanical unloading. Eur J Heart Fail 2010;12:444-53.
36. Gruson D, Ahn SA, Ketelslegers JM, Rousseau MF. Increased plasma myostatin in heart failure. Eur J Heart Fail 2011;13:734-6.
37. Lima AR, Martinez PF, Okoshi K, et al. Myostatin and follistatin expression in skeletal muscles of rats with chronic heart failure. Int J Exp Pathol 2010;91:54-62.
38. Rock KL, Gramm C, Rothstein L, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 1994;78:761-71.
40. Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 2013;6:25-39.
41. Schiaffino S, Hanzlíkovávěra. Studies on the effect of denervation in developing muscle. II. The lysosomal system. J Ultrastruct Res 1972;39:1-14.
42. Furuno K, Goodman MN, Goldberg AL. Role of different proteolytic systems in the degradation of muscle proteins during denervation atrophy. J Biol Chem 1990;265:8550-7.
43. García-Prat L, Martínez-Vicente M, Perdiguero E, et al. Autophagy maintains stemness by preventing senescence. Nature 2016;529:37-42.
44. Sousa-Victor P, Gutarra S, García-Prat L, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014;506:316-21.
45. Deval C, Mordier S, Obled C, et al. Identification of cathepsin L as a differentially expressed message associated with skeletal muscle wasting. Biochem J 2001;360:143.
46. Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007;6:458-71.
47. Dobrowolny G, Aucello M, Rizzuto E, et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab 2008;8:425-36.
48. Lecker SH, Jagoe RT, Gilbert A, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 2004;18:39-51.
49. Adams V, Jiang H, Yu J, et al. Apoptosis in skeletal myocytes of patients with chronic heart failure is associated with exercise intolerance. J Am College Cardiol 1999;33:959-65.
50. Vescovo G, Volterrani M, Zennaro R, et al. Apoptosis in the skeletal muscle of patients with heart failure: investigation of clinical and biochemical changes. Heart 2000;84:431-7.
51. Libera LD, Zennaro R, Sandri M, Ambrosio GB, Vescovo G. Apoptosis and atrophy in rat slow skeletal muscles in chronic heart failure. Am J Physiol 1999;277:C982-6.
52. Mitchell RG, Duscha BD, Robbins JL, et al. Increased levels of apoptosis in gastrocnemius skeletal muscle in patients with peripheral arterial disease. Vasc Med 2007;12:285-90.
53. Schaap LA, Pluijm SM, Deeg DJ, et al. Higher inflammatory marker levels in older persons: associations with 5-year change in muscle mass and muscle strength. J Gerontol A Biol Sci Med Sci 2009;64:1183-9.
54. Tuttle CSL, Thang LAN, Maier AB. Markers of inflammation and their association with muscle strength and mass: a systematic review and meta-analysis. Ageing Res Rev 2020;64:101185.
55. Dalla Libera L, Sabbadini R, Renken C, et al. Apoptosis in the skeletal muscle of rats with heart failure is associated with increased serum levels of TNF-alpha and sphingosine. J Mol Cell Cardiol 2001;33:1871-8.
56. Okutsu M, Call JA, Lira VA, et al. Extracellular superoxide dismutase ameliorates skeletal muscle abnormalities, cachexia, and exercise intolerance in mice with congestive heart failure. Circ Heart Fail 2014;7:519-30.
57. Allard MF, Schönekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol 1994;267:H742-50.
58. Zhu Q, Gao R, Zhang Y, et al. Dysbiosis signatures of gut microbiota in coronary artery disease. Physiol Genomics 2018;50:893-903.
59. Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol 2021;19:55-71.
60. Tang WH, Wang Z, Levison BS, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 2013;368:1575-84.
61. Luedde M, Winkler T, Heinsen FA, et al. Heart failure is associated with depletion of core intestinal microbiota. ESC Heart Fail 2017;4:282-90.
62. Beale AL, O’Donnell JA, Nakai ME, et al. The gut microbiome of heart failure with preserved ejection fraction. J Am Heart Assoc 2021;10:e020654.
63. Tang WHW, Li DY, Hazen SL. Dietary metabolism, the gut microbiome, and heart failure. Nat Rev Cardiol 2019;16:137-54.
64. Niebauer J, Volk H, Kemp M, et al. Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet 1999;353:1838-42.
65. Buffa JA, Romano KA, Copeland MF, et al. The microbial gbu gene cluster links cardiovascular disease risk associated with red meat consumption to microbiota L-carnitine catabolism. Nat Microbiol 2022;7:73-86.
66. Koeth RA, Wang Z, Levison BS, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 2013;19:576-85.
67. Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011;472:57-63.
68. Zhu W, Gregory JC, Org E, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 2016;165:111-24.
69. Tan Y, Zhou J, Liu C, et al. Association between plasma trimethylamine n-oxide and neoatherosclerosis in patients with very late stent thrombosis. Can J Cardiol 2020;36:1252-60.
70. Tan Y, Sheng Z, Zhou P, et al. Plasma trimethylamine N-oxide as a novel biomarker for plaque rupture in patients with ST-segment-elevation myocardial infarction. Circ Cardiovasc Interv 2019;12:e007281.
71. Li XS, Obeid S, Klingenberg R, et al. Gut microbiota-dependent trimethylamine N-oxide in acute coronary syndromes: a prognostic marker for incident cardiovascular events beyond traditional risk factors. Eur Heart J 2017;38:814-24.
72. Li XS, Wang Z, Cajka T, et al. Untargeted metabolomics identifies trimethyllysine, a TMAO-producing nutrient precursor, as a predictor of incident cardiovascular disease risk. JCI Insight 2018;3:99096.
73. Al-Obaide MAI, Singh R, Datta P, et al. Gut microbiota-dependent trimethylamine-N-oxide and serum biomarkers in patients with T2DM and advanced CKD. J Clin Med 2017;6:86.
74. Chen S, Henderson A, Petriello MC, et al. Trimethylamine N-oxide binds and activates perk to promote metabolic dysfunction. Cell Metab 2019;30:1141-1151.e5.
75. Ufnal M, Jazwiec R, Dadlez M, Drapala A, Sikora M, Skrzypecki J. Trimethylamine-N-oxide: a carnitine-derived metabolite that prolongs the hypertensive effect of angiotensin II in rats. Can J Cardiol 2014;30:1700-5.
76. Li T, Chen Y, Gua C, Li X. Elevated circulating trimethylamine N-oxide levels contribute to endothelial dysfunction in aged rats through vascular inflammation and oxidative stress. Front Physiol 2017;8:350.
77. Brunt VE, Gioscia-Ryan RA, Casso AG, et al. Trimethylamine-N-Oxide promotes age-related vascular oxidative stress and endothelial dysfunction in mice and healthy humans. Hypertension 2020;76:101-12.
78. Krauss RM, Eckel RH, Howard B, et al. AHA dietary guidelines: revision 2000: a statement for healthcare professionals from the nutrition committee of the american heart association. Circulation 2000;102:2284-99.
79. Topping DL, Clifton PM. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev 2001;81:1031-64.
81. Brown AJ, Goldsworthy SM, Barnes AA, et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem 2003;278:11312-9.
82. Kaisar MMM, Pelgrom LR, van der Ham AJ, Yazdanbakhsh M, Everts B. Butyrate conditions human dendritic cells to prime type 1 regulatory t cells via both histone deacetylase inhibition and g protein-coupled receptor 109a signaling. Front Immunol 2017;8:1429.
83. Pluznick JL, Protzko RJ, Gevorgyan H, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci USA 2013;110:4410-5.
84. Ang Z, Er JZ, Tan NS, et al. Human and mouse monocytes display distinct signalling and cytokine profiles upon stimulation with FFAR2/FFAR3 short-chain fatty acid receptor agonists. Sci Rep 2016;6:34145.
85. Masui R, Sasaki M, Funaki Y, et al. G protein-coupled receptor 43 moderates gut inflammation through cytokine regulation from mononuclear cells. Inflamm Bowel Dis 2013;19:2848-56.
86. Huang W, Man Y, Gao C, et al. Short-chain fatty acids ameliorate diabetic nephropathy via GPR43-mediated inhibition of oxidative stress and NF-. κ ;2020:4074832.
87. Lee SU, In HJ, Kwon MS, et al. β-Arrestin 2 mediates G protein-coupled receptor 43 signals to nuclear factor-κB. Biol Pharm Bull 2013;36:1754-9.
88. Marques FZ, Nelson E, Chu PY, et al. High-Fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation 2017;135:964-77.
89. Zhang L, Deng M, Lu A, et al. Sodium butyrate attenuates angiotensin II-induced cardiac hypertrophy by inhibiting COX2/PGE2 pathway via a HDAC5/HDAC6-dependent mechanism. J Cell Mol Med 2019;23:8139-50.
90. Liu CF, Tang WHW. Epigenetics in cardiac hypertrophy and heart failure. JACC Basic Transl Sci 2019;4:976-93.
91. Li M, van Esch BCAM, Henricks PAJ, Folkerts G, Garssen J. The Anti-inflammatory Effects of short chain fatty acids on lipopolysaccharide- or tumor necrosis factor α-stimulated endothelial cells via activation of GPR41/43 and Inhibition of HDACs. Front Pharmacol 2018;9:533.
92. Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci USA 2014;111:2247-52.
93. Brown JM, Hazen SL. The gut microbial endocrine organ: bacterially derived signals driving cardiometabolic diseases. Annu Rev Med 2015;66:343-59.
94. Astrup A, Magkos F, Bier DM, et al. Saturated fats and health: a reassessment and proposal for food-based recommendations: JACC state-of-the-art review. J Am Coll Cardiol 2020;76:844-57.
95. Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem 2003;72:137-74.
96. Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res 2006;47:241-59.
97. Staels B, Fonseca VA. Bile acids and metabolic regulation: mechanisms and clinical responses to bile acid sequestration. Diabetes Care 2009;32 Suppl 2:S237-45.
98. Watanabe M, Houten SM, Mataki C, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006;439:484-9.
99. Binah O, Rubinstein I, Bomzon A, Better OS. Effects of bile acids on ventricular muscle contraction and electrophysiological properties: studies in rat papillary muscle and isolated ventricular myocytes. Naunyn Schmiedebergs Arch Pharmacol 1987;335:160-5.
100. Bogin E, Better O, Harari I. The effect of jaundiced sera and bile salts on cultured beating rat heart cells. Experientia 1983;39:1307-8.
101. Sheikh Abdul Kadir SH, Miragoli M, Abu-Hayyeh S, et al. Bile acid-induced arrhythmia is mediated by muscarinic M2 receptors in neonatal rat cardiomyocytes. PLoS One 2010;5:e9689.
102. Williamson C, Gorelik J, Eaton BM, Lab M, Swiet MD, Korchev Y. The bile acid taurocholate impairs rat cardiomyocyte function: a proposed mechanism for intra-uterine fetal death in obstetric cholestasis. Clin Sci 2001;100:363-9.
103. Gorelik J, Harding SE, Shevchuk AI, et al. Taurocholate induces changes in rat cardiomyocyte contraction and calcium dynamics. Clin Sci (Lond) 2002;103:191-200.
104. Desai MS, Mathur B, Eblimit Z, et al. Bile acid excess induces cardiomyopathy and metabolic dysfunctions in the heart. Hepatology 2017;65:189-201.
105. Mayerhofer CCK, Ueland T, Broch K, et al. Increased secondary/primary bile acid ratio in chronic heart failure. J Card Fail 2017;23:666-71.
106. Ebner N, Anker SD, von Haehling S. Recent developments in the field of cachexia, sarcopenia, and muscle wasting: highlights from the 11th Cachexia Conference. J Cachexia Sarcopenia Muscle 2019;10:218-25.
107. Toney AM, Fox D, Chaidez V, Ramer-Tait AE, Chung S. Immunomodulatory role of urolithin a on metabolic diseases. Biomedicines 2021;9:192.
108. Zanotti I, Dall’Asta M, Mena P, et al. Atheroprotective effects of (poly)phenols: a focus on cell cholesterol metabolism. Food Funct 2015;6:13-31.
109. García-Mantrana I, Calatayud M, Romo-Vaquero M, Espín JC, Selma MV, Collado MC. Urolithin metabotypes can determine the modulation of gut microbiota in healthy individuals by tracking walnuts consumption over three days. Nutrients 2019;11:2483.
110. Savi M, Bocchi L, Mena P, et al. In vivo administration of urolithin A and B prevents the occurrence of cardiac dysfunction in streptozotocin-induced diabetic rats. Cardiovasc Diabetol 2017;16:80.
111. Tang L, Mo Y, Li Y, et al. Urolithin A alleviates myocardial ischemia/reperfusion injury via PI3K/Akt pathway. Biochem Biophys Res Commun 2017;486:774-80.
112. Ghosh N, Das A, Biswas N, et al. Urolithin A augments angiogenic pathways in skeletal muscle by bolstering NAD+ and SIRT1. Sci Rep 2020;10:20184.
113. Nemet I, Saha PP, Gupta N, et al. A Cardiovascular disease-linked gut microbial metabolite acts via adrenergic receptors. Cell 2020;180:862-877.e22.
114. Melamed ML, Plantinga L, Shafi T, et al. Retained organic solutes, patient characteristics and all-cause and cardiovascular mortality in hemodialysis: results from the retained organic solutes and clinical outcomes (ROSCO) investigators. BMC Nephrol 2013;14:134.
115. Wu IW, Hsu KH, Hsu HJ, et al. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients--a prospective cohort study. Nephrol Dial Transplant 2012;27:1169-75.
116. Lin CJ, Pan CF, Liu HL, et al. The role of protein-bound uremic toxins on peripheral artery disease and vascular access failure in patients on hemodialysis. Atherosclerosis 2012;225:173-9.
117. Huang ST, Shu KH, Cheng CH, et al. Serum total p-cresol and indoxyl sulfate correlated with stage of chronic kidney disease in renal transplant recipients. Transplant Proc 2012;44:621-4.
118. Lekawanvijit S, Adrahtas A, Kelly DJ, Kompa AR, Wang BH, Krum H. Does indoxyl sulfate, a uraemic toxin, have direct effects on cardiac fibroblasts and myocytes? Eur Heart J 2010;31:1771-9.
119. Yisireyili M, Shimizu H, Saito S, Enomoto A, Nishijima F, Niwa T. Indoxyl sulfate promotes cardiac fibrosis with enhanced oxidative stress in hypertensive rats. Life Sci 2013;92:1180-5.
120. Peng YS, Ding HC, Lin YT, Syu JP, Chen Y, Wang SM. Uremic toxin p-cresol induces disassembly of gap junctions of cardiomyocytes. Toxicology 2012;302:11-7.
121. Han H, Zhu J, Zhu Z, et al. p-Cresyl sulfate aggravates cardiac dysfunction associated with chronic kidney disease by enhancing apoptosis of cardiomyocytes. J Am Heart Assoc 2015;4:e001852.
122. Six I, Flissi N, Lenglet G, et al. Uremic toxins and vascular dysfunction. Toxins (Basel) 2020;12:404.
123. Lv J, Chen J, Wang M, Yan F. Klotho alleviates indoxyl sulfate-induced heart failure and kidney damage by promoting M2 macrophage polarization. Aging (Albany NY) 2020;12:9139-50.
124. Chen K, Wang S, Sun QW, Zhang B, Ullah M, Sun Z. Klotho deficiency causes heart aging via impairing the Nrf2-GR pathway. Circ Res 2021;128:492-507.
125. Siddharth J, Chakrabarti A, Pannérec A, et al. Aging and sarcopenia associate with specific interactions between gut microbes, serum biomarkers and host physiology in rats. Aging (Albany NY) 2017;9:1698-720.
126. Bindels LB, Beck R, Schakman O, et al. Restoring specific lactobacilli levels decreases inflammation and muscle atrophy markers in an acute leukemia mouse model. PLoS One 2012;7:e37971.
127. Munukka E, Rintala A, Toivonen R, et al. Faecalibacterium prausnitzii treatment improves hepatic health and reduces adipose tissue inflammation in high-fat fed mice. ISME J 2017;11:1667-79.
128. Varian BJ, Goureshetti S, Poutahidis T, et al. Beneficial bacteria inhibit cachexia. Oncotarget 2016;7:11803-16.
129. Walsh ME, Bhattacharya A, Sataranatarajan K, et al. The histone deacetylase inhibitor butyrate improves metabolism and reduces muscle atrophy during aging. Aging Cell 2015;14:957-70.
130. Wu CS, Wei Q, Wang H, et al. Protective effects of ghrelin on fasting-induced muscle atrophy in aging mice. J Gerontol A Biol Sci Med Sci 2020;75:621-30.
131. Lustgarten MS. The Role of the gut microbiome on skeletal muscle mass and physical function: 2019 update. Front Physiol 2019;10:1435.
132. Abrigo J, Campos F, Gonzalez F, et al. Sarcopenia Induced by chronic liver disease in mice requires the expression of the bile acids membrane receptor TGR5. Int J Mol Sci 2020;21:7922.
133. Thibaut MM, Sboarina M, Roumain M, et al. Inflammation-induced cholestasis in cancer cachexia. J Cachexia Sarcopenia Muscle 2021;12:70-90.
134. He W, Luo Y, Liu J, et al. Trimethylamine N-Oxide, a gut microbiota-dependent metabolite, is associated with frailty in older adults with cardiovascular disease. CIA 2020;15:1809-20.
135. Enoki Y, Watanabe H, Arake R, et al. Indoxyl sulfate potentiates skeletal muscle atrophy by inducing the oxidative stress-mediated expression of myostatin and atrogin-1. Sci Rep 2016;6:32084.
136. Rodrigues GGC, Dellê H, Brito RBO, et al. Indoxyl sulfate contributes to uremic sarcopenia by inducing apoptosis in myoblasts. Arch Med Res 2020;51:21-9.
137. Sato E, Mori T, Mishima E, et al. Metabolic alterations by indoxyl sulfate in skeletal muscle induce uremic sarcopenia in chronic kidney disease. Sci Rep 2016;6:36618.
138. Picca A, Ponziani FR, Calvani R, et al. Gut microbial, inflammatory and metabolic signatures in older people with physical frailty and sarcopenia: results from the biosphere study. Nutrients 2019;12:65.
139. Buigues C, Fernández-Garrido J, Pruimboom L, et al. Effect of a prebiotic formulation on frailty syndrome: a randomized, double-blind clinical trial. Int J Mol Sci 2016;17:932.
140. Aoyagi Y, Amamoto R, Park S, et al. Independent and interactive effects of habitually ingesting fermented milk products containing lactobacillus casei strain shirota and of engaging in moderate habitual daily physical activity on the intestinal health of older people. Front Microbiol 2019;10:1477.
141. Ferrari R, Cargnoni A, Ceconi C. Anti-ischaemic effect of ivabradine. Pharmacol Res 2006;53:435-9.
142. Griffiths EJ, Rutter GA. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim Biophys Acta 2009;1787:1324-33.
143. Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 2012;13:566-78.
144. Coen PM, Musci RV, Hinkley JM, Miller BF. Mitochondria as a Target for Mitigating Sarcopenia. Front Physiol 2018;9:1883.
145. Chaudhary KR, El-Sikhry H, Seubert JM. Mitochondria and the aging heart. J Geriatr Cardiol 2011;8:159-67.
146. Gonzalez-Freire M, Scalzo P, D’Agostino J, et al. Skeletal muscle ex vivo mitochondrial respiration parallels decline in vivo oxidative capacity, cardiorespiratory fitness, and muscle strength: The Baltimore Longitudinal Study of Aging. Aging Cell 2018;17:e12725.
147. Lai L, Leone TC, Keller MP, et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail 2014;7:1022-31.
148. Tezze C, Romanello V, Desbats MA, et al. Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab 2017;25:1374-1389.e6.
149. Sebastián D, Sorianello E, Segalés J, et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J 2016;35:1677-93.
150. Leduc-Gaudet JP, Hussain SNA, Barreiro E, Gouspillou G. Mitochondrial dynamics and mitophagy in skeletal muscle health and aging. Int J Mol Sci 2021;22:8179.
151. Casuso RA, Huertas JR. The emerging role of skeletal muscle mitochondrial dynamics in exercise and ageing. Ageing Res Rev 2020;58:101025.
152. Li YP, Chen Y, Li AS, Reid MB. Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am J Physiol Cell Physiol 2003;285:C806-12.
153. Aucello M, Dobrowolny G, Musaro A. Localized accumulation of oxidative stress causes muscle atrophy through activation of an autophagic pathway. Autophagy 2009;5:527-9.
154. Romanello V, Guadagnin E, Gomes L, et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J 2010;29:1774-85.
155. Salminen A, Ojala J, Kaarniranta K, Kauppinen A. Mitochondrial dysfunction and oxidative stress activate inflammasomes: impact on the aging process and age-related diseases. Cell Mol Life Sci 2012;69:2999-3013.
156. Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev 2004;18:357-68.
157. Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest 2006;116:615-22.
158. Arany Z, He H, Lin J, et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab 2005;1:259-71.
159. Viña J, Gomez-Cabrera MC, Borras C, et al. Mitochondrial biogenesis in exercise and in ageing. Adv Drug Deliv Rev 2009;61:1369-74.
160. Dillon LM, Rebelo AP, Moraes CT. The role of PGC-1 coactivators in aging skeletal muscle and heart. IUBMB Life 2012;64:231-41.
161. Migliavacca E, Tay SKH, Patel HP, et al. Mitochondrial oxidative capacity and NAD+ biosynthesis are reduced in human sarcopenia across ethnicities. Nat Commun 2019;10:5808.
162. Kong X, Wang R, Xue Y, et al. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One 2010;5:e11707.
163. Joseph AM, Adhihetty PJ, Buford TW, et al. The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell 2012;11:801-9.
164. Liu HW, Chang YC, Chan YC, Hu SH, Liu MY, Chang SJ. Dysregulations of mitochondrial quality control and autophagic flux at an early age lead to progression of sarcopenia in SAMP8 mice. Biogerontology 2020;21:367-80.
165. Lahiri S, Kim H, Garcia-Perez I, et al. The gut microbiota influences skeletal muscle mass and function in mice. Sci Transl Med 2019;11:eaan5662.
166. Ryu D, Mouchiroud L, Andreux PA, et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med 2016;22:879-88.
167. Makrecka-Kuka M, Volska K, Antone U, et al. Trimethylamine N-oxide impairs pyruvate and fatty acid oxidation in cardiac mitochondria. Toxicol Lett 2017;267:32-8.
168. Vaiserman AM, Koliada AK, Marotta F. Gut microbiota: A player in aging and a target for anti-aging intervention. Ageing Res Rev 2017;35:36-45.
169. Buford TW. (Dis)Trust your gut: the gut microbiome in age-related inflammation, health, and disease. Microbiome 2017;5:80.
170. Harber MP, Konopka AR, Undem MK, et al. Aerobic exercise training induces skeletal muscle hypertrophy and age-dependent adaptations in myofiber function in young and older men. J Appl Physiol (1985) 2012;113:1495-504.
171. Konopka AR, Harber MP. Skeletal muscle hypertrophy after aerobic exercise training. Exerc Sport Sci Rev 2014;42:53-61.
172. Bori Z, Zhao Z, Koltai E, et al. The effects of aging, physical training, and a single bout of exercise on mitochondrial protein expression in human skeletal muscle. Exp Gerontol 2012;47:417-24.
173. Johnston AP, De Lisio M, Parise G. Resistance training, sarcopenia, and the mitochondrial theory of aging. Appl Physiol Nutr Metab 2008;33:191-9.
174. Clarke SF, Murphy EF, O’Sullivan O, et al. Exercise and associated dietary extremes impact on gut microbial diversity. Gut 2014;63:1913-20.
175. Taniguchi H, Tanisawa K, Sun X, et al. Effects of short-term endurance exercise on gut microbiota in elderly men. Physiol Rep 2018;6:e13935.
176. Munukka E, Ahtiainen JP, Puigbó P, et al. Six-week endurance exercise alters gut metagenome that is not reflected in systemic metabolism in over-weight women. Front Microbiol 2018;9:2323.
177. Šoltys K, Lendvorský L, Hric I, et al. Strenuous physical training, physical fitness, body composition and bacteroides to prevotella ratio in the gut of elderly athletes. Front Physiol 2021;12:670989.
178. Perry CA, Gadde KM. The role of calorie restriction in the prevention of cardiovascular disease. Curr Atheroscler Rep 2022;24:235-42.
179. Li L, Su Y, Li F, et al. The effects of daily fasting hours on shaping gut microbiota in mice. BMC Microbiol 2020;20:65.
180. van der Merwe M, Sharma S, Caldwell JL, et al. Time of feeding alters obesity-associated parameters and gut bacterial communities, but not fungal populations, in C57BL/6 male mice. Curr Dev Nutr 2020;4:nzz145.
181. Beli E. Restructuring of the gut microbiome by intermittent fasting prevents retinopathy and prolongs survival in db/db mice. Diabetes 2018;67:1867-79.
182. Cignarella F, Cantoni C, Ghezzi L, et al. Intermittent Fasting Confers Protection in CNS Autoimmunity by Altering the Gut Microbiota. Cell Metab 2018;27:1222-1235.e6.
183. Fraumene C. Caloric restriction promotes rapid expansion and long-lasting increase of Lactobacillus in the rat fecal microbiota. Gut Microbes 2018;9:104-114.
184. Nagata S, Asahara T, Ohta T, et al. Effect of the continuous intake of probiotic-fermented milk containing Lactobacillus casei strain Shirota on fever in a mass outbreak of norovirus gastroenteritis and the faecal microflora in a health service facility for the aged. Br J Nutr 2011;106:549-56.
185. Akatsu H, Iwabuchi N, Xiao JZ, et al. Clinical effects of probiotic Bifidobacterium longum BB536 on immune function and intestinal microbiota in elderly patients receiving enteral tube feeding. JPEN J Parenter Enteral Nutr 2013;37:631-40.
186. Ostan R, Béné MC, Spazzafumo L, et al. Impact of diet and nutraceutical supplementation on inflammation in elderly people. Results from the RISTOMED study, an open-label randomized control trial. Clin Nutr 2016;35:812-8.
187. Nagata S, Asahara T, Wang C, et al. The Effectiveness of lactobacillus beverages in controlling infections among the residents of an aged care facility: a randomized placebo-controlled double-blind trial. Ann Nutr Metab 2016;68:51-9.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Liu CF, Tang WHW. Gut microbiota in sarcopenia and heart failure. J Cardiovasc Aging 2022;2:35. http://dx.doi.org/10.20517/jca.2022.07
AMA Style
Liu CF, Tang WHW. Gut microbiota in sarcopenia and heart failure. The Journal of Cardiovascular Aging. 2022; 2(3): 35. http://dx.doi.org/10.20517/jca.2022.07
Chicago/Turabian Style
Liu, Chia-Feng, W. H. Wilson Tang. 2022. "Gut microbiota in sarcopenia and heart failure" The Journal of Cardiovascular Aging. 2, no.3: 35. http://dx.doi.org/10.20517/jca.2022.07
ACS Style
Liu, C.F.; Tang WHW. Gut microbiota in sarcopenia and heart failure. J. Cardiovasc. Aging. 2022, 2, 35. http://dx.doi.org/10.20517/jca.2022.07
About This Article
Copyright

Data & Comments
Data


 Cite This Article 16 clicks
Cite This Article 16 clicks

 Like This Article 14
likes
Like This Article 14
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.