
GSK-3β mediates cardiac senescence through inhibition of ULK1 directed autophagy
 0
0Abstract
Cardiac senescence is the progressive decline in cardiac performance resulting from decline in age related structural and metabolic processes. Aging cardiac myocytes exhibit alterations in fatty acid and glucose oxidation metabolism, activation of innate immune signaling, enhanced fibrosis, mitochondrial dysfunction, endoplasmic reticulum stress, DNA damage, senescence-associated secreting phenotype and impaired autophagy. GSK-3β is an upstream regulator of autophagy through its interaction with ULK1 which regulates the initiation step of autophagy and formation of the autophagosme. Herein, we highlight a novel putative molecular mechanism that functionally links cardiac senescence, hypertrophy and autophagy regulation. Ser9 phosphorylation of GSK-3β is critical for promoting cardiac senescence via reduction in ULK1 phosphorylation at Ser913 and inhibition of autophagy.
Keywords
Cardiac senescence is defined as the progressive decline in cardiac function resulting from deterioration of structural and metabolic processes associated with advanced age. At the cellular level, alterations in fatty acid and glucose oxidation, innate immune signaling, and fibrosis are common underlying features of aging and, over time, profoundly influence ventricular remodeling and cardiac performance. In addition, at the molecular level, aged cardiac myocytes exhibit organellar defects that include mitochondrial dysfunction, endoplasmic reticulum stress, DNA damage, and senescence-associated secreting phenotype (SASP)[1].
Recent evidence has shown that autophagy, which is crucial for maintaining cellular and tissue homeostasis by removing damaged proteins and organelles from cells, is functionally impaired with age. Autophagy consists of several sequential steps: initiation, nucleation, elongation, and fusion with lysosomes. The initiation of autophagy includes the formation of the autophagosome, which is regulated by Unc-51 like autophagy activating kinase 1 (ULK1). ULK1 is a serine/threonine kinase that phosphorylates Beclin-1, subsequently leading to the phosphorylation and activation of the Vps34 kinase complex, which gives rise to the autophagosome. Autophagy-related genes allow the autophagosomes to mature and subsequently fuse with the lysosome-associated soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) to form the autophago-lysosome. Hence, the inhibition or activation of ULK1 can profoundly influence autophagy and the clearance of cellular debris - identifying ULK1 as a critical regulator of the autophagic process. While positive activation of ULK1 is regulated by AMP-activated protein kinase, it is negatively regulated by the mechanistic target of rapamycin (mTOR). mTOR complex 1 (mTORC1), which is comprised of mTOR and regulatory-associated protein of mTOR (Raptor), phosphorylate and inhibit ULK1 kinase activity, thereby inhibiting autophagy[2]. Therefore, the decline in cardiac autophagy with aging, results in the accumulation of misfolded proteins and dysfunctional organelles such as mitochondria which impair cardiac function. Consequently, studies have shown that while inhibition of autophagy exacerbates pathologies associated with cardiac senescence, activation of autophagy can restore cardiac function and ameliorate aging-related cardiac dysfunction[3].
Glycogen synthase kinase-3β (GSK-3β) is a serine/threonine kinase that has a role in regulating autophagy in cardiomyocytes. GSK-3β is active under basal conditions and is inhibited during stress conditions. Inhibition of GSK-3β is achieved through phosphorylation in Ser9. One of the principal kinases that phosphorylate GSK-3β at Ser9 is Akt/PI3K, which is phosphorylated and activated by mTORC2[4]. Previously, GSK-3β was reported as an upstream regulator of autophagy in adult hippocampal neural stem cells through its interaction with ULK1 via phosphorylation of Ser405 and Ser415 within the GABARAP-interacting region[5]. In the heart, GSK-3β was shown to be activated during ischemia but inhibited during reperfusion, suggesting that adaptive activation of GSK-3β during ischemia stimulates autophagy, whereas inhibition of GSK-3β inactivates autophagy during reperfusion. Inhibiting GSK-3β expression during both ischemia and reperfusion was detrimental to cardiac function[4].
Notably, GSK-3β also plays an essential role in senescence. Phosphorylation of GSK-3β and its subsequent inactivation was reported in aged hepatocytes[6]. Notably, the GSK-3β promoter was suppressed by C/EBPβ-histone deacetylase 1 complexes in aging hepatocytes, resulting in reduced GSK-3β levels and loss of the liver’s regenerative capacity[7]. In the context of the heart, phosphorylation levels of both Akt and GSK-3β were increased during aging, together with the aging-associated increase in mitochondrial permeability pore opening (mPTP)[8]. During cardiac hypertrophy, another sine qua non of cardiac senescence, elevated levels of GSK-3β were shown to protect against hypertrophic cardiac myocyte growth. This negative effect of GSK-3β on cardiac hypertrophy may be attributed to transcriptional inhibition of proteins such as β-catenin, nuclear factor of activated T cells, GATA Binding Protein 4 (GATA4), and eukaryotic initiation factor 2Be. This observation is supported by the fact that overexpression of wild-type GSK-3β or knock-in mutations of Ser9 GSK-3β (GSK-3βS9A) protected the heart from cardiac hypertrophy. Similarly, inhibition of GSK-3β with lithium stimulated the hypertrophic response. Further evidence to support a role for
A new study by Chen et al.[10] in this issue of The Journal of Cardiovascular Aging provides molecular insight into the underlying mechanisms that functionally link cardiac aging, hypertrophy, and autophagy regulation. Through various elegantly designed studies, the authors show that Ser9 phosphorylation of GSK-3β promotes cardiac senescence via the reduction in ULK1 phosphorylation at Ser913 and inhibition of autophagy.
To better understand the underlying mechanisms of GSK-3β mediated cardiac senescence, Chen et al.[10] first assessed the levels of total and Ser9 phosphorylated GSK-3β in mouse hearts at different ages. Notably, the levels of Ser9 phosphorylated GSK-3β were increased in the hearts of 24 months old mice, compared to the hearts of mice at 6 months of age. This finding was further supported by the observed increased levels of GSK-3β substrates, such as β-catenin and myeloid cell leukemia 1 in the 24 months old mice, which are known to be increased when GSK-3β is inactivated[11].
Next, the authors examined how the increased phosphorylation of GSK-3β at Ser9 affects cardiac function and survival rates of wild-type (WT) mice and mice in which a mutant form of GSK-3β defective for serine 9 phosphorylation (GSK-3βS9A) was knocked-in (βKI) in germ-line. As expected, βKI mice showed higher survival rates than WT mice and coincided with improved cardiac function in the βKI mice. Further, while both systolic and diastolic dysfunction were developed in 24 months old WT mice, the βKI mice were found to be protected from age-related cardiac dysfunction, suggesting that increased Ser9 phosphorylated GSK-3β plays a pivotal role in controlling systolic and diastolic cardiac function during aging. In addition, other hallmarks associated with cardiac senescence, including cardiac hypertrophy, fibrosis, apoptosis, and SASP, were significantly increased in WT mice and abrogated in the βKI mice. These results highlighted a critical role for Ser9 phosphorylated GSK-3β in promoting senescence in the heart.
Previous studies have shown that autophagy is dramatically decreased with aging, contributing to age-related pathologies in the heart[1]. The findings by Chen et al.[10] support this notion by demonstrating reduced LC3II levels and increased p62 levels in hearts of 24 months old WT mice indicating autophagy impairment. In contrast, βKI mice showed higher levels of LC3II and lower levels of p62, suggesting that autophagy was active. Interestingly, overexpression of WT GSK-3β increased LC3II and reduced p62 and stimulated expression of GFP-LC3 puncta, indicating that GSK-3β can activate autophagy in cardiac myocytes.
While studying the molecular mechanism by which GSK-3β controls autophagy, Chen et al.[10] identified that ULK1 has a consensus amino acid sequence for GSK-3β-mediated phosphorylation. To validate that ULK1 was a putative phosphorylation target for GSK-3β, the authors performed a kinase assay using recombinant GSK-3β and a kinase-inactive mutant of ULK1 (ULK1K46R). The findings of this assay showed that GSK-3β could directly phosphorylate ULK1. The phosphorylation site in mice was also verified by mass spectrometry, which identified Ser913 as the key serine on ULK1 targeted by GSK-3β. Recognizing this specific phosphorylation site led the authors to generate ULK1S913A knock-in mouse. Interestingly, the ULK1S913A mice showed higher levels of p62 and reduced levels of LC3-II, compared to WT mice, demonstrating that phosphorylation of ULK1 at Ser913 plays a pivotal role in regulating GSK-3β-induced activation of autophagy in the heart.
While this study provides compelling new evidence that ULK1 phosphorylation at Ser913 by GSK-3β is an essential step for initiation of autophagy during cardiac aging, it also leaves a few open questions that need to be further considered. One question that arises from the study is what happens to the upstream regulators of GSK-3β, such as Akt/PI3K and mTORC1/2, during aging? Are these pathways influenced in
Another crucial question that should be considered arises from a recent study that demonstrated that ULK1 regulates the initiation step of autophagy and the final step of fusion with lysosome and syntaxin 17[12]. Since Chen et al.[10] assessed autophagy activation and autophagy flux only, it remains unclear whether ULK1 phosphorylation at Ser913 by GSK-3β affects the last step of fusion with lysosome as well. Furthermore, the same study showed that phosphorylation of ULK1 enhanced its interaction with heat shock cognate 70 kDa protein (HSC70), therefore increasing its degradation through chaperone-mediated autophagy (CMA)[12]. It is intriguing to test whether the mechanism by which GSK-3β regulates autophagy through ULK1 phosphorylation can stimulate CMA also or mainly macroautophagy.
Nevertheless, the study by Chen et al.[10] has revealed a novel and important signaling pathway for autophagy activation during cardiac senescence through ULK1 phosphorylation at Ser913 by GSK-3β, Figure 1. Addressing the critical signaling pathways and molecular effectors that link autophagy activation by ULK1 may prove beneficial in establishing how modulating autophagy regulation can be applied clinically to curtail cardiac dysfunction and hypertrophy during the inevitable evolution of aging and senescence.
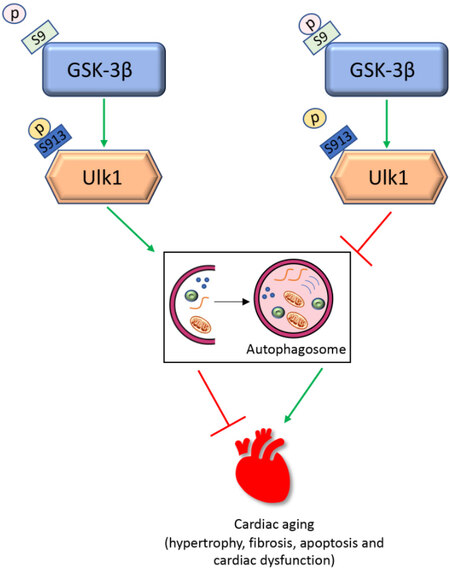
Figure 1. A scheme representing GSK-3β mediated cardiac senescence through inhibition of ULK-1 directed autophagy. Under basal conditions, phosphorylation of ULK1 at S913 by GSK-3β promotes autophagy. During aging, phosphorylation of GSK-3β at S9 prevents activation of ULK1, leading to inhibition of autophagy and stimulation of cardiac aging processes, including hypertrophy, fibrosis, apoptosis, and cardiac dysfunction.
DECLARATIONS
Authors’ contributionsCollected the data, wrote the paper and revised it: Rabinovich-Nikitin I
Wrote the paper and revised it: Kirshenbaum LA
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by a Foundation grant to Kirshenbaum LA from the Canadian Institute for Health Research (CIHR) and St. Boniface Hospital Foundation, Kirshenbaum LA holds a Canada Research Chair in Molecular Cardiology; Rabinovich-Nikitin I holds a Post-doctoral Fellowship from the CIHR.
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Tang X, Li PH, Chen HZ. Cardiomyocyte senescence and cellular communications within myocardial microenvironments. Front Endocrinol (Lausanne) 2020;11:280.
2. Wong PM, Puente C, Ganley IG, Jiang X. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 2013;9:124-37.
3. Dutta D, Calvani R, Bernabei R, Leeuwenburgh C, Marzetti E. Contribution of impaired mitochondrial autophagy to cardiac aging: mechanisms and therapeutic opportunities. Circ Res 2012;110:1125-38.
4. Zhai P, Sadoshima J. Glycogen synthase kinase-3β controls autophagy during myocardial ischemia and reperfusion. Autophagy 2012;8:138-9.
5. Ryu HY, Kim LE, Jeong H, et al. GSK3B induces autophagy by phosphorylating ULK1. Exp Mol Med 2021;53:369-83.
6. Souder DC, Anderson RM. An expanding GSK3 network: implications for aging research. Geroscience 2019;41:369-82.
7. Jin J, Wang GL, Shi X, Darlington GJ, Timchenko NA. The age-associated decline of glycogen synthase kinase 3beta plays a critical role in the inhibition of liver regeneration. Mol Cell Biol 2009;29:3867-80.
8. Zhu J, Rebecchi MJ, Tan M, Glass PS, Brink PR, Liu L. Age-associated differences in activation of Akt/GSK-3beta signaling pathways and inhibition of mitochondrial permeability transition pore opening in the rat heart. J Gerontol A Biol Sci Med Sci 2010;65:611-9.
9. Sugden PH, Fuller SJ, Weiss SC, Clerk A. Glycogen synthase kinase 3 (GSK3) in the heart: a point of integration in hypertrophic signalling and a therapeutic target? Br J Pharmacol 2008;153 Suppl 1:S137-53.
10. Chen Y, Maejima Y, Shirakabe A, et al. Ser9 phosphorylation of GSK-3β promotes aging in the heart through suppression of autophagy. J Cardiovasc Aging 2021;1:9.
11. Hirotani S, Zhai P, Tomita H, et al. Inhibition of glycogen synthase kinase 3beta during heart failure is protective. Circ Res 2007;101:1164-74.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Rabinovich-Nikitin I, Kirshenbaum LA. GSK-3β mediates cardiac senescence through inhibition of ULK1 directed autophagy. J Cardiovasc Aging 2021;1:14. http://dx.doi.org/10.20517/jca.2021.24
AMA Style
Rabinovich-Nikitin I, Kirshenbaum LA. GSK-3β mediates cardiac senescence through inhibition of ULK1 directed autophagy. The Journal of Cardiovascular Aging. 2021; 1(1): 14. http://dx.doi.org/10.20517/jca.2021.24
Chicago/Turabian Style
Rabinovich-Nikitin, Inna, Lorrie A. Kirshenbaum. 2021. "GSK-3β mediates cardiac senescence through inhibition of ULK1 directed autophagy" The Journal of Cardiovascular Aging. 1, no.1: 14. http://dx.doi.org/10.20517/jca.2021.24
ACS Style
Rabinovich-Nikitin, I.; Kirshenbaum LA. GSK-3β mediates cardiac senescence through inhibition of ULK1 directed autophagy. J. Cardiovasc. Aging. 2021, 1, 14. http://dx.doi.org/10.20517/jca.2021.24
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 10 clicks
Cite This Article 10 clicks

 Like This Article 31
likes
Like This Article 31
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.