
S-nitrosoglutathione reductase (GSNOR) deficiency accelerates cardiomyocyte differentiation of induced pluripotent stem cells
 0
0Abstract
Introduction: Induced pluripotent stem cells (iPSCs) provide a model of cardiomyocyte (CM) maturation. Nitric oxide signaling promotes CM differentiation and maturation, although the mechanisms remain controversial.
Aim: The study tested the hypothesis that in the absence of S-nitrosoglutathione reductase (GSNOR), a denitrosylase regulating protein S-nitrosylation, the resultant increased S-nitrosylation accelerates the differentiation and maturation of iPSC-derived cardiomyocytes (CMs).
Methods and Results: iPSCs derived from mice lacking GSNOR (iPSCGSNOR-/-) matured faster than wildtype iPSCs (iPSCWT) and demonstrated transient increases in expression of murine Snail Family Transcriptional Repressor 1 gene (Snail), murine Snail Family Transcriptional Repressor 2 gene (Slug) and murine Twist Family BHLH Transcription Factor 1 gene (Twist), transcription factors that promote epithelial-to-mesenchymal transition (EMT) and that are regulated by Glycogen Synthase Kinase 3 Beta (GSK3β). Murine Glycogen Synthase Kinase 3 Beta
Conclusion: Together these findings suggest that increased S-nitrosylation of Gsk3β promotes CM differentiation and maturation from iPSCs. Manipulating the post-translational modification of GSK3β may provide an important translational target and offers new insight into understanding of CM differentiation from pluripotent stem cells.
One sentence summary: Deficiency of GSNOR or addition of GSNO accelerates early differentiation and maturation of iPSC-cardiomyocytes.
Keywords
INTRODUCTION
The maturation of cardiomyocytes (CM) derived from induced pluripotent (iPS) cells is a highly complex yet incompletely understood process. Endogenous and exogenous nitric oxide (NO) affects stem cell biology and promotes CM maturation, although the mechanism(s) remains controversial[1,2]. S-nitrosylation (S-NO) of cysteine thiols is a major signaling pathway through which NO exerts its broad regulatory actions[3,4]. Levels of S-NO in mice are physiologically regulated via the activity of S-nitrosoglutathione (GSNO) reductase [GSNOR (alcohol dehydrogenase 5)], an enzyme that promotes protein denitrosylation. Mice lacking GSNOR (GSNOR-/- mice) show increased levels of S-NO proteins[5-7]. GSNOR-/- mice have enhanced cardiomyocyte proliferation and recover better than wildtype mice post-myocardial infarction[8,9]. Here, we hypothesized that the absence of GSNOR, and the resultant increased S-nitrosylation, accelerates the differentiation and maturation of cardiomyocytes. We analyzed early cardiomyogenic differentiation of iPS cells derived from GSNOR-/- and wildtype [WT (control)] mice to test this hypothesis.
Colony structural integrity is crucial for maintaining pluripotency during the expansion of induced pluripotent stem cells (iPSCs) and requires that cells do not undergo epithelial-to-mesenchymal transition (EMT). EMT is a highly ordered process that generates pools of primitive stem/progenitor cells at precise locations within the embryo[10]. EMT is initiated by the downregulation of intercellular adhesion proteins, such as E-cadherin, the prototypical epithelial adhesion protein, and by the upregulation of EMT-inducing transcription factors, such as Snail (encoded by SNAI1 and SNAI2)[11]. Glycogen Synthase Kinase 3 Beta (GSK3β), a serine/threonine kinase, plays a central role in controlling Snail Family Transcriptional Repressor 1 (Snail) expression and the Wnt/β-catenin pathway, an important initiator of EMT[12]. Modulation of the GSK and Wnt pathways (with small-molecule inhibitors) affects the CM maturation of iPSCs[13,14]. S-NO of GSK3β reduces its activity[15], and inhibition of GSK3β activity promotes cardiac differentiation and proliferation in murine and human PSCs[16].
Here, for the first time, we show that GSNOR loss of function promotes EMT, S-NO of GSK3β, and CM proliferation and differentiation and accelerates the maturation of iPSC-CMs. These findings provide new insights into the modulation of iPSC-cardiomyogenesis and potential cardiac repair targets and cell-based therapy.
MATERIAL AND METHODS
This study was reviewed and approved by the University of Miami Institutional Animal Care and Use Committee (Protocol #20-168) and complies with all federal and state guidelines concerning animals in research and teaching as defined by “The Guide for the Care and Use of Laboratory Animals”[17].
Animals
The generation of GSNOR-/- mice and their backcrossing for > 10 generations into a C57Bl6/J mouse background (WT) to eliminate/minimize genetic heterogeneity has been reported[8]. Age-matched WT (C57Bl6/J) mice obtained from Jackson Laboratories were used as controls. Mice received food and water ad libitum and were on a 12-h light/dark cycle. At the endpoint, mice were euthanized by either cervical dislocation or isoflurane inhalation, followed by exsanguination.
Generation of iPSCs
Murine iPSCs derived from GSNOR-/- mice (iPSCGSNOR-/-) and iPSCs derived from the wild type mice (iPSCWT) were generated from fibroblasts isolated from adult heart of 3.5-mo-old male GSNOR-/- and WT mice, respectively, using a commercially available kit (STEMCCA, Millipore). Briefly, hearts were minced and fibroblasts at passage one plated on gelatin-coated 12-well plates at a density of 2 × 104 cells per well (Day 0) with Dulbecco’s modified eagle media (DMEM) (Gibco), 2 mM l-glutamine, 10% (vol/vol) fetal bovine serum (FBS) (ES-Gibco), and 1% penicillin-streptomycin (Gibco). The next day (Day 1), cells were transduced with lentiviral particles (STEMCCA cassette). The STEMCCA cassette is a humanized excisable system containing all four reprogramming factors POU Class 5 Homeobox 1, SRY-Box Transcription Factor 2 (SOX2) Kruppel Like Factor 4, and c-MYC in a single “stem cell cassette” (pHAGE2-EF1aFull-hOct4-F2A-hKlf4-IRES-hSox2-P2A-hcMyc-W-loxP). The reprogramming Kit was used at a multiplicity of infection (MOI) of 100, followed by a repeated infection at an MOI of 75 on Day 2. Approximately 48 h later, cells were collected by trypsinization and replated onto fresh plates coated with irradiated mouse embryonic fibroblasts (MEFs; Millipore) and fed with mouse ES medium [DMEM, 2 mM l-glutamine, 20% (vol/vol) FBS (ES-Gibco), 0.1 mM nonessential amino acids (Gibco), 0.1 mM 2-mercaptoethanol (Gibco), and 1,000 units/mL Leukemia inhibitory factor (LIF) (Millipore)] supplemented with a cocktail of small molecules to enhance reprogramming (Mouse iPS reprogramming Boost Supplement, Millipore). iPSC colonies began to emerge on Day 7. Each colony was manually picked and individually transferred to fresh plates coated with MEFs. A total of 12 iPSCGSNOR-/- and 10 iPSCWT clones were generated. Clones 9 and 11 for iPSCGSNOR-/- and clones 6 and 10 for iPSCWT were selected and expanded for this study. No variability was observed between studied clones. All iPSC lines were grown on MEFs and adapted gradually to a modified ES + 2i medium [DMEM, l-glutamine, 20% (vol/vol) knockout serum replacement (Gibco), 0.1 mM nonessential amino acids, 0.1 mM 2-mercaptoethanol, 1000 units/mL LIF, 1 µM PD0325901 (Tocris) and 3 µM CHIR99021 (Tocris)]. The human induced pluripotent stem cell (hiPSC) line SC101A (System Biosciences) was maintained in E8 medium (ThermoFisher Scientific) under feeder-free conditions on Matrigel-coated plates (BD Biosciences). Cardiac differentiation of human pluripotent stem cells (hPSCs) was performed according to the previously described protocol for directed differentiation via small molecule modulation[18].
GSNOR activity in whole heart lysates and iPSCs
The GSNOR activity assay was performed as previously described[8]. Briefly, heart tissues from WT and GSNOR-/- male mice were minced/homogenized in a cell lysis buffer (Cell Signaling). The protein concentration of these samples was determined using a standard Bradford assay. Heart homogenate
Gene-expression analysis
Total RNA was extracted from iPSCGSNOR-/-, iPSCWT and human induced pluripotent stem cells (hiPSCs) at selected time points before and during their differentiation into cardiomyocytes, using the RNeasy mini plus kit, according to the manufacturers’ instructions (Qiagen). cDNA synthesis was performed using the high-capacity cDNA reverse-transcription kit, according to the manufacturer’s instructions (Applied Biosystems). Quantitative PCR was performed using Taqman Universal Master mix in an iQ5 real-time PCR (qRT-PCR) detection system (Bio Rad). All samples were run in triplicates and normalized to a GAPDH/18s endogenous control. Relative fold-change was calculated using the ΔΔCt method. In addition, Taqman Gene-expression assays were performed for the following murine genes: t, alcohol Dehydrogenase 5 (Class III) (Adh5), Snail1, Snail2, E-cadherin, POU Class 5 Homeobox gene (Oct4), Sox2, Kruppel Like Factor 4, murine gene (Klf4), Glycogen Synthase Kinase 3 Beta gene (Gsk3β), ATPase Sarcoplasmic/Endoplasmic Reticulum Ca2+ Transporter 2, murine gene (Atp2a2), Ryanodine Receptor 2
MTT assay
MTT assays were carried out on Day 4 to 5 of the differentiation process. Cell viability was measured by using Vybrant® MTT Cell Proliferation Assay Kit (Invitrogen by ThermoFisher Scientific) according to the manufacturer’s protocol (Quick Protocol Option). Cells were plated in 24-well plates at the density of 2.5 × 104 cells/well. Briefly, the cells were labeled with MTT and incubated for 4 h at 37 ℃. Then, DMSO was added to each well and incubated at 37 ℃ for 10 min to dissolve the formazan. Absorbance at a wavelength of 540 nm was measured in a Spectromax M5 spectrophotometer (Spectra Max; Molecular Devices, Sunnyvale, CA, USA) using software: SoftMaxPro 6.5.1.
Bromodeoxyuridine proliferation assay
Bromodeoxyuridine (BrdU) is incorporated into the newly synthesized DNA strands of actively proliferating cells. According to the manufacturer’s protocol, we determined cell proliferation using a BrdU Cell Proliferation enzyme-linked immunosorbent assay kit (Abcam, ab126556, USA). Briefly, 10, 20, or 30 embryoid bodies (EBs) were seeded in a 96-well plate and received BrdU (20 μL). After 3 (Day 5) or 24 (Day 4-5) h of incubation (37 ℃, 5% CO2), cells were fixed, permeabilized and the DNA denatured. Next, Anti-BrdU monoclonal antibodies were added and incubated for 1 hour. Absorbance was then obtained using a Spectromax M5 spectrophotometer (Spectra Max; Molecular Devices, Sunnyvale, CA, USA) at a wavelength of 450 nm.
Beating cells
Video files were captured using a Zeiss Axio Vert.A1 inverted microscope under phase contrast. The resulting video file was then processed into digitized representations of moving pixels, after which the sum of moving pixels was plotted vs. time. Finally, 10-s segments of these videos were employed for the visual plots.
Migration assay
Cell migration was detected using the Transwell system (Corning, Inc., Corning, NY, USA). Cells (1.5 × 105) were trypsinized and seeded into the upper chamber. After incubation for 24 h at 37 ℃, the cells that had migrated into the lower compartment were fixed with methanol, stained with a 5% crystal violet solution, and counted. Each experiment was conducted with at least four replicates, and migrated cells were counted in a minimum of 3 microscope fields.
Western blotting
For Western blotting, cell lysates were prepared by re-suspending iPSC-derived embryoid bodies in RIPA lysis buffer (R0278, Sigma) and protease inhibitor (Complete protease inhibitor cocktail; Roche Molecular Biochemicals). Proteins (25-40 μg/lane) were electrophoresed through 8%-10% polyacrylamide-SDS gel and then transferred to the nitrocellulose membrane. Membranes were immunostained overnight at 4 ℃ with primary Axin 1 (1:1000, Cell Signaling Technology, #2087), Gsnor (1:1000, Proteintech, 11051-1-AP), Hsp90 (1:1000, Cell Signaling Technology, #4874), T-brachyury (1:1000, Abcam, ab20680) antibodies and
Resin-assisted capture of S-nitrosothiols (SNO-RAC) assay
S-nitrosothiols (SNO)-RAC assays were performed in the dark as described[19]. Briefly, iPSCs were homogenized in HEN buffer (250 mM HEPES, 1 mM EDTA, and 0.1 mM neocuproine, pH 7.7). Free cysteine residues were blocked with MMTS and reacted with or without sodium ascorbate. Protein lysate was incubated with thiol-reactive resin (Sigma-Aldrich) for 5 h. Resin-captured proteins were eluted using 50 μL elution buffer (20 mM HEPES, 100 mM NaCl, 1 mM EDTA, 100 mM β-mercaptoethanol) and heated at 95 ℃ for 5 min in reducing SDS-PAGE loading buffer. The expression of murine Glycogen Synthase Kinase 3 Beta protein (Gsk3β) was determined by Western blot analysis.
Cytosolic calcium measurement
Intracellular Ca2+ was measured using the Ca2+-sensitive dye Fura-2 and a dual-excitation spectrofluorometer (IonOptix LLC, Milton, MA, USA). Briefly, cells were loaded with 2.5 μM Fura-2 for 20 min at room temperature in Tyrode’s buffer containing (in mM): 144 NaCl, 1 MgCl2, 10 HEPES, 5.6 glucose, 5 KCl, 1.2 NaH2PO4, 1.5 CaCl2 (adjusted to a pH 7.4 with NaOH). Then the cells were washed with fresh regular Tyrode’s solution for at least 10 min. Cells were excited with a xenon lamp at 340 nm and
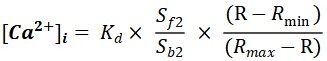
Kd (dissociation constant) in adult myocytes was taken as 224 nmol/L. The scaling factors Sf2 and Sb2 were extracted from calibration[3].
Calcium transient kinetics and sarcoplasmic reticulum function
We measured the time to reach the (Ca2+) transient peak and the time of decaying from the peak to 50% over baseline (tt50%). We also assessed the time constant (τ) of Ca2+ decay from a regular twitch and after a pulse of 20 mM caffeine to block the sarcoplasmic reticulum (SR) re-uptake both in regular conditions (1.5 mM CaCl2) and in the absence of Na+ and Ca2+ to block the Na+/Ca2+ exchanger (NCX) activity as well. Using these parameters, we calculated the proportional contribution of sarco(endo)plasmic reticulum Ca2+-ATPase, human protein (SERCA), NCX, and other non-NCX mechanisms in mouse induced pluripotent stem cells (miPSCs)-derived CMs by comparing the time constants of Ca2+ decay in the following conditions:
• Regular twitch 1/τtwitch = KSERCA + KNCX + Knon-NCX
• Caffeine challenge (1.5 mM CaCl2) 1/τCaff = KNCX + Knon-NCX
• Caffeine challenge (0Na+/0Ca2+) 1/τCaff0Na/0Ca = Knon-NCX
where K represent the time constant for each depicted mechanism.
Statistics
Statistical analyses were performed using GraphPad Prism version 9.00 for Mac. Values were analyzed using Student’s t-test or two-way ANOVA. All data met the assumptions of the tests. Therefore, a P < 0.05 was considered statistically significant. All values are reported as mean ± SEM (n is noted in the figures).
RESULTS
S-nitrosylation status is linked to early cardiomyogenesis in murine iPSCs
Nitric oxide synthase inhibitors prevent the maturation of mESCs into differentiated cardiomyocytes, a process that can be readily rescued by NO donors[20]. To investigate the effect of NO-mediated protein S-nitrosylation during early cardiomyogenesis in our miPSC model, we used an NADH-dependent GSNOR activity assay to gauge GSNOR enzymatic activity in GSNOR-/- and C57Bl6/J wild type (WT) minced adult heart. As expected, in the presence of GSNO, NADH oxidation produced a sharp decrease in absorbance in WT but not in GSNOR-/- lysates over time [Figure 1A]. Murine iPSCGSNOR-/- and iPSCWT from these adult heart-minced fibroblasts were generated after virus infection using the OSKM factors protocol[21]. Spontaneous differentiation of iPSCs into beating cardiomyocytes subsequent to the formation of 3-dimensional EBs was seen, as depicted in the schematic experimental approach [Figure 1B]. EBs were grown in hanging drops for the first two days and neither Gsnor gene expression nor protein levels were observed in the iPSCGSNOR-/- cells on Day 0 to Day 6 of the differentiation process [Figure 1C and D]. In addition, to provide some characterization on the pluripotency of our cells, we observed an accelerated loss of pluripotency markers such as Oct4, Sox2 and Klf4 in both groups, but, in the iPSCGSNOR-/- group on Day 6, Oct4 and Sox2 were significantly lower than control iPSCs. In contrast, Klf4 expression, which is also involved in proliferation and differentiation[22], was higher in iPSCGSNOR-/- on Day 2 but not at later timepoints [Figure 1E].
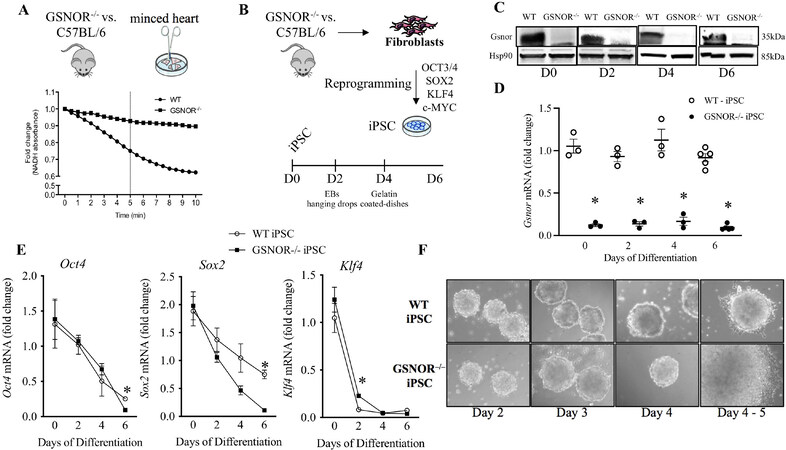
Figure 1. GSNOR loss of function accelerates cardiomyocyte differentiation of murine iPSCs. (A) GSNOR activity in whole heart lysates of GSNOR-/- and C57BL/6 mice. (B) Schematic representation of cardiomyocyte differentiation procedure from Day 0 to Day 6. (C) Western blot analysis of Gsnor and Hsp90 (internal control) in murine iPSCWT and iPSCGSNOR-/- during cardiomyocyte differentiation. (D) qRT-PCR analysis of Gsnor in murine iPSCWT and iPSCGSNOR-/- during cardiomyocyte differentiation. (E) qRT-PCR analysis of pluripotent genes (Oct4, Sox2, Klf4) in murine iPSCWT (open circles) and iPSCGSNOR-/- (black boxes) during cardiomyocyte differentiation. (F) Morphology of embryoid bodies from iPSCWT (top) and iPSCGSNOR-/- (bottom) over a time course of 5 days. Data are the mean ± SEM of 3 independent experiments. P-values were calculated using Student’s t-test. *P < 0.05.
Finally, the morphology of EB differentiation was examined over a period of 6 days (Day 0-6). The formation and maintenance of the EB population remained relatively uniform until Day 4, at which point, we observed continuous proliferation/differentiation and apparent EMT only in the iPSCGSNOR-/-. Thus, we demonstrated that the genetic deletion of GSNOR is directly associated with differences at the early stages of miPSC growth.
GSNOR deletion stimulates cell proliferation and migration in murine iPSCs
NO, as described above, can influence the proliferation and differentiation cascades of certain cell types[23,24]. Indeed, S-nitrosylation may be integral to NO function during a variety of cellular processes. We previously described that GSNOR-/- mice exhibit the enhanced proliferative activity of adult cardiac progenitors and cardiomyocytes post-myocardial infarction[9]. Therefore, we next examined the effect of GSNOR deletion on cell proliferation and migration in miPSCs. An MTT assay was performed on Day 5 and showed that iPSCGSNOR-/- have a higher proliferative (metabolic) activity compared to iPSCWT (0.096 ± 0.006 vs. 0.12 ± 0.006; P < 0.05) [Figure 2A]. We observed enhanced BrdU incorporation in iPSCGSNOR-/- EBs (20 and 30 EBs/well) compared with WT [Figure 2B and C], in agreement with the results of the MTT assay, which also indicated increased proliferation. We also investigated if the absence of GSNOR activity promotes cell migration in addition to proliferation. To quantify migration, cells were seeded into the upper chamber of a transwell dish and after 24 h of incubation; cells that migrated were counted and stained with crystal violet solution. We observed that cellular migration increased by 75% in iPSCGSNOR-/- compared with iPSCWT [Figure 2D], 3.3 ± 0.16 vs. 5.8 ± 0.3; P < 0.05). Representative micrographs at Day 4 are shown in Figure 2E. These results demonstrate that iPSCGSNOR-/- exhibit increased cellular migration and proliferation compared to iPSCWT.
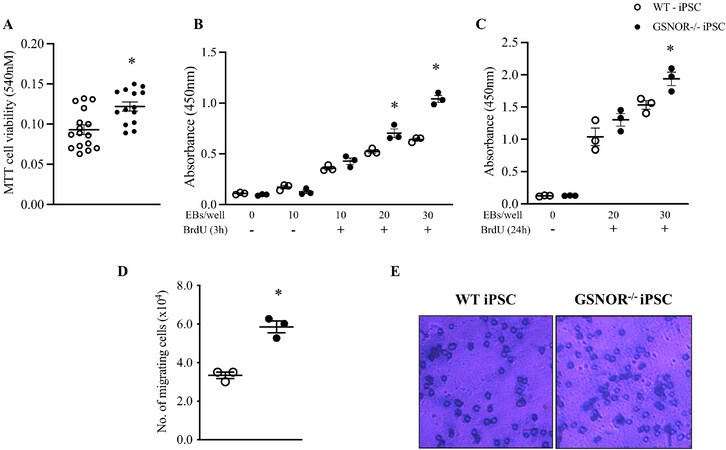
Figure 2. GSNOR loss of function stimulates cell proliferation and cell migration in murine iPSCs. (A) Cell viability as measured by MTT assay on Day 4 in murine iPSCGSNOR-/- and iPSCWT. EBs (4000-5000 cells/EB) from murine iPSCWT (white circles) and iPSCGSNOR-/- (black circles) were incubated with BrdU and proliferation determined following short- (3 h, B), and long-term (24 h, C) plating of different numbers of EBs (4000-5000 cells/EB). (D) Average number of murine iPSCGSNOR-/- and iPSCWT that migrated over a 24-h period. E, Micrograph of cells that migrated and then stained with crystal violet. Data are the mean ± SEM of 3 independent experiments. P-values were calculated using Student’s t-test at each time point. *P < 0.05.
GSNOR deletion reduces Gsk3β and increase Axin 1 and Axin 2 in iPSCGSNOR-/-
As mentioned above, S-nitrosylation of GSK3β inhibits its kinase activity independent of the canonical phospho-inhibition pathway[15]. Furthermore, inhibition of GSK3 is sufficient to induce CM cell cycle re-entry, DNA synthesis, and likely proliferation[16]. We hypothesized that GSNOR loss of function increases GSK3β S-nitrosylation and decreases Gsk3β expression/functionality during the initial growth and differentiation of iPSCs. To test this hypothesis, we first measured mRNA levels of Gsk3β from Day 0 to Day 6. iPSCGSNOR-/- showed significantly reduced expression levels of Gsk3β on Day 4 compared to iPSCWT [Figure 3A]. We next performed SNO-RAC assays[19] on iPSCWT and iPSCGSNOR-/-. S-nitrosylated proteins were captured by thiol-reactive resin, and Gsk3β S-nitrosylation status was assessed by immunoblotting. iPSCGSNOR-/- demonstrated greater S-nitrosylation of Gsk3β compared to iPSCWT [Figure 3B]. The identification of endogenous S-NO in the SNO-RAC assay was validated by the omission of ascorbate, which prevents S-nitrosothiols (SNOs) from being reduced. Furthermore, we examined if increased NO levels in iPSCWT, mimic the effects of the suppressed Gsk3β levels seen in iPSCGSNOR-/-. For these experiments, we examined three different concentrations of the NO donor, GSNO, 25 µM, 125 µM and 250 µM for 3 h on Day 4 of differentiation. We observed that GSNO decreased Gsk3β expression in iPSCWT cells by 40% [Figure 3C]. The same experiment was performed in hiPSC using two different concentrations of GSNO (100 µM and 250 µM) for 24 h on Day 2 of cell culture, a time point that appears to be equivalent to murine iPSC on Day 4[25]; human Glycogen Synthase Kinase 3 Beta gene (GSK3β) expression was reduced by ~50% with 100 μM GSNO (P < 0.05) compared to control [Figure 3D]. These studies demonstrated that increased S-nitrosylation or exposure to a pharmacologic NO donor reduces GSK3β expression in iPSCs.
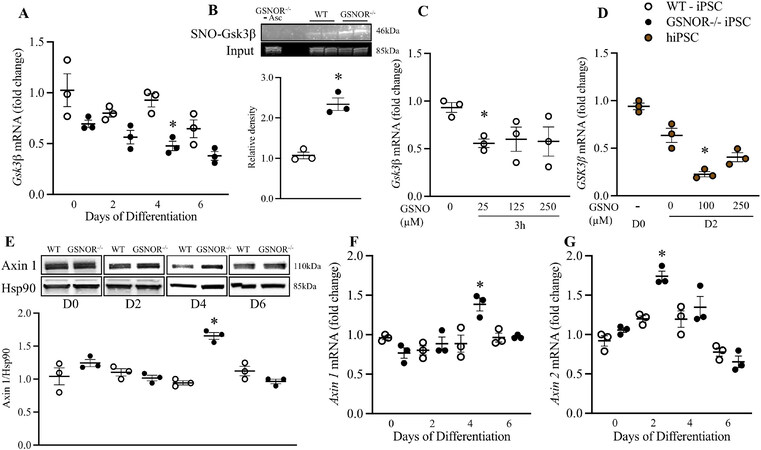
Figure 3. GSNOR loss of function alters S-nitrosylation and expression of GSK3β. (A) qRT-PCR analysis of Gsk3β in murine iPSCWT (white circles) and iPSCGSNOR-/- (black circles) during cardiomyocyte differentiation. (B) S-nitrosylation of Gsk3β in murine iPSCGSNOR-/- and iPSCWT as assessed by SNO-RAC on Day 4. Effects on Gsk3β mRNA levels, in (C) murine iPSCWT, following 3 h incubation with 3 different concentrations of the NO donor, GSNO, on Day 4, and in (D) hiPSCs (brown circles), following 24 h of incubation with GSNO beginning on Day 1. (E) Western blot analysis of Axin 1 and Hsp90 (internal control) in murine iPSCWT (white circles) and iPSCGSNOR-/- (black circles) on Day 0, 2, 4 and 6 of cardiomyocyte differentiation. qRT-PCR analysis of Axin 1 (F) and Axin 2 (G) in murine iPSCWT (white circles) and iPSCGSNOR-/- (black circles) during cardiomyocyte differentiation. Data are the mean ± SEM of 3 independent experiments. P-values were calculated using Student’s t-test at each time point. *P < 0.05.
Axin, a scaffold protein, that forms a complex with GSK3β and β-catenin and promotes GSK3β-dependent phosphorylation of β-catenin, thereby stimulating the degradation of β-catenin. Axin 1 and Axin 2 share a number of structural and functional properties, including homologous GSK3β-binding domains. Surprisingly, Axin 1 protein and mRNA expression on Day 4 [Figure 3D and E], and Axin 2 mRNA expression, on Day 2 were increased in iPSCGSNOR-/- compared to iPSCWT [Figure 3F]. These results suggest that aberrant S-NO/NO levels impact multiple components of the β-catenin regulatory pathway.
EMT-related transcription factors twist, slug, and snail are upregulated, and E-cadherin is downregulated in murine iPSCGSNOR-/- and in hiPSCs treated with GSNO
Previously described, inhibition of GSK3β stimulates the transcription of Snail, a repressor of E-cadherin and inducer of EMT[26]. Similar cooperative activities suggest that Twist and Snail work synergistically to induce EMT[27]. Since migration, proliferation and EMT are closely related processes, we tested whether GSNOR loss of function, affects EMT-related transcription factors in murine and human iPSCs. qRT-PCR studies revealed upregulation of Snail 1 (Day 4), murine Snail Family Transcriptional Repressor 2 gene
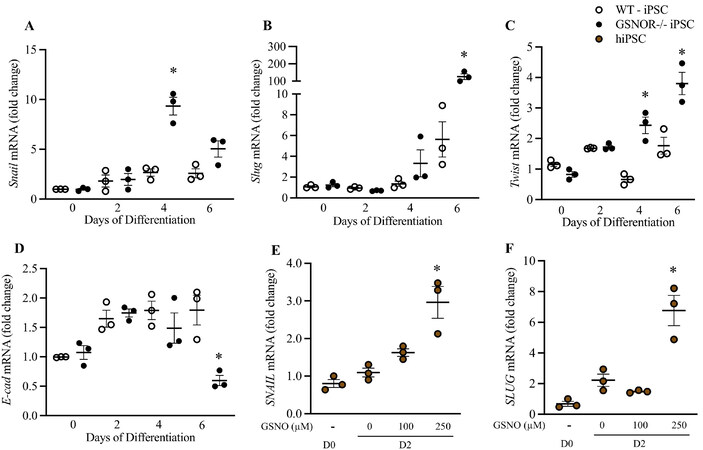
Figure 4. GSNOR loss of function regulates EMT-Related transcription factors in murine iPSCGSNOR-/- and hiPSCs treated with GSNO. qRT-PCR analysis of Snail (A), Slug (B), Twist (C) and E-cadherin (D) in murine iPSCWT (white circles) and iPSCGSNOR-/- (black circles) during cardiomyocyte differentiation. qRT-PCR analysis of SNAIL (E) and SLUG (F) in hiPSC, on Day 2, following 24 h incubation with GSNO (brown circles). Data are the mean ± SEM of 3 independent experiments. P-values were calculated using Student’s t-test. *P < 0.05.
GSNOR deletion reduces Brachyury (T) and promotes early cardiac differentiation
Multiple upstream signals regulate the mesoderm marker, brachyuryexpression including Phosphatidylinositol-3 kinase (PI3K)/Akt pathway[28] and GSK3β via the Wnt/β-catenin pathway[29]. Recent work suggests that brachyury is essential for specifying cell fate but dispensable for cell survival, proliferation and EMT[30]. Thus, we investigated the effects of altered S-nitrosylation on the function of brachyury in our iPSC model. Surprisingly, brachyury mRNA expression was reduced on Day 4 in
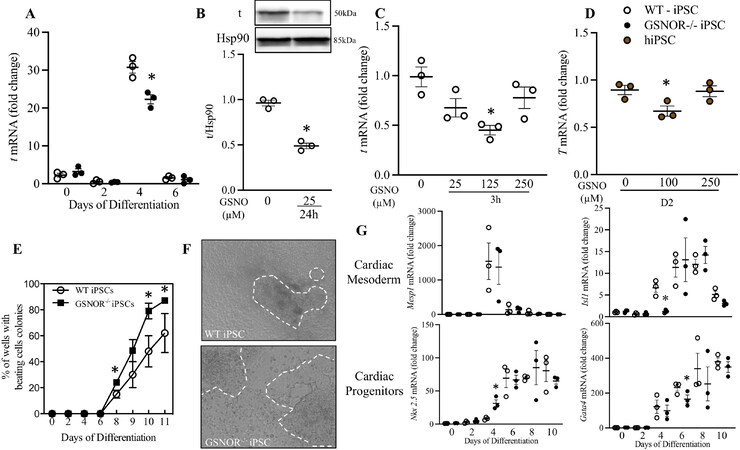
Figure 5. GSNOR loss of function reduces Brachyury (T) and accelerates cardiomyocyte differentiation. (A) qRT-PCR analysis of t in murine iPSCWT (white circles) and iPSCGSNOR-/- (black circles) during cardiomyocyte differentiation. WB (B) and qRT-PCR (C) of t in murine iPSCWT on Day 4, following 24 h of incubation with GSNO. (D) qRT-PCR analysis of T in hiPSC, on Day 2, following 24 h incubation with GSNO. (E) Percentage of beating colonies from murine iPSCWT and iPSCGSNOR-/- during cardiomyocyte differentiation. (F) Representative images of organoids from murine iPSCWT (top) iPSCGSNOR-/- (bottom) on Day 10 of cardiomyocyte differentiation. (G) qRT-PCR analysis of Cardiac mesoderm (Mesp1, Isl1) and Cardiac Progenitors genes (Nkx 2.5, Gata4) in murine iPSCWT and iPSCGSNOR-/- during cardiomyocyte differentiation. Data are the mean ± SEM of 3 independent experiments. P-values were calculated using Student’s t-test. *P < 0.05.
iPSCGSNOR-/- exhibited an increased percentage of beating EBs on Days 8, 10, and 11. On Day 10, 80% of iPSCGSNOR-/- EB outgrowth area displayed beating compared with 48% of iPSCWT EBs [Figure 5E]. Representative images of organoids [Figure 5F] and video of beating organoids was captured by standard brightfield microscopy (Supplementary Movie 1 - beating organoids of iPSCWT; Supplementary Movie 2 - beating organoids of iPSCGSNOR-/-). In addition to brachyury, mRNA for stage-specific markers of cardiac mesoderm (Isl1) and cardiac progenitors (Nkx 2.5 and Gata4) were also altered at specific timepoints in iPSCGSNOR-/- compared to iPSCWT. Isl1 expression was decreased and Nkx 2.5 was significantly increased on Day 4 while Gata4 expression was decreased on Day 6 [Figure 5G]. Mesp1 expression was not different between the two iPSCs [Figure 5G]. Together, these results demonstrate that increased NO bioavailability in iPSCGSNOR-/- promotes a faster transition from mesoderm to cardiac progenitor, as measured by increasing Nkx 2.5 expression.
GSNOR deletion impacts cardiomyocyte structural maturation
As CM differentiation progresses, there are specific isoform switches of structural proteins indicative of the maturity of differentiating CMs, e.g., increased myosin heavy chain MYH6/MYH7 and troponin I ssTNI/TNNi1 to cTNI/TNNi3 expression ratios, and the emergence of genes involved in calcium handling, such as RyR2, sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) and Na+/Ca2+ exchanger (NCX)[31]. To understand the maturation of our undifferentiated iPSCs into cardiomyocytes, and to assess the impact of S-nitrosylation on this process, we investigated the expression of these CM-related genes on Days 10 and 21 of differentiation/maturation. Tnni1 expression was increased on Day 10 in iPSCGSNOR-/- compared to iPSCWT. Tnni3 was detected at the early stage (Day 0), followed by a sharp decrease and a gradual increase thereafter. For this gene, we observed a significant increase in iPSCGSNOR-/- on Day 4 and Day 10 [Figure 6A]. Immunofluorescence analyses confirmed our previous results that troponin I expression increased on Day 10 in the iPSCGSNOR-/- [Figure 6B]. Following typical differentiation protocols, the sarcomeres of hPSC-CMs are usually composed of a mixture of α (MYH6) and β (MYH7) isoforms of the myosin heavy chain[32]. In our study, both Myh6 or Myh7 expression were significantly increased in iPSCGSNOR-/--CMs compared to iPSCWT on Days 8 and 10 [Figure 6A]. We evaluated the expression of markers of cardiomyocyte maturation, including structural proteins, with increasing differentiation time (Day 21). Tnni1, Tnni3, Myh6, and Myh7 were significantly increased in iPSCGSNOR-/--CMs compared to iPSCWT-CMs on Day 21 [Figure 6C]. We next evaluated the presence and relative expression of the mature calcium-handling molecular components, RyR2 and Atp2a2, in miPSC-CMs. Both genes were expressed at significantly higher levels in iPSCGSNOR-/- compared to iPSCWT. We also observed that the sarcomere appeared better organized in iPSCGSNOR-/- on Day 21 as revealed by α-actinin staining [Figure 6D]. These results of structural proteins, calcium handling genes, and sarcomeric α-actinin suggest that, at this stage of cardiomyocyte differentiation of iPSCGSNOR-/- is more advanced than iPSCWT.
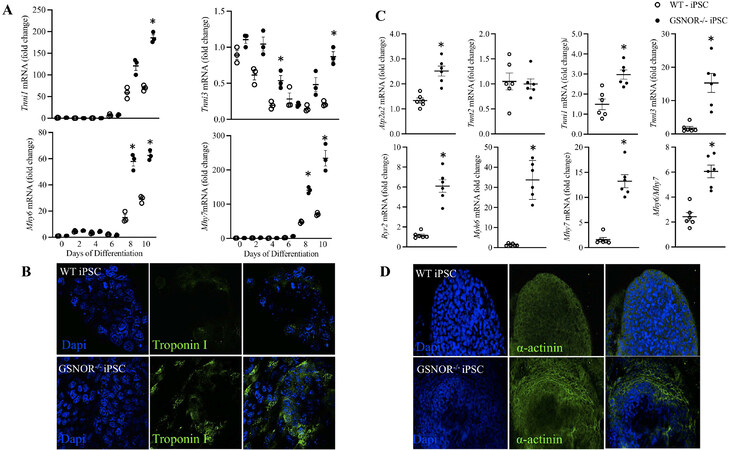
Figure 6. GSNOR loss of function impacts cardiomyocyte structural maturation. (A) qRT-PCR analysis of Cardiac genes, troponin I1 and I3 (Tnni1, Tnni3), myosin heavy chain isoforms-6 (Myh6) and -7 (Myh7) in murine iPSCWT (white circles) and iPSCGSNOR-/- (black circles) during cardiomyocyte differentiation. (B) Representative immunofluorescence staining of Troponin I in outgrowth of beating EBs on Day 10 of murine iPSCWT (top) iPSCGSNOR-/- (bottom). (C) qRT-PCR analysis of Myocardial genes associated with calcium (Ca2+) handling and cardiac contraction/relaxation, including sarco/endoplasmic reticulum Ca2+ ATPase 2 [sarco(endo)plasmic reticulum Ca2+-ATPase, murine gene (Serca)2], troponin T2 (Tnnt2), Tnni1, Tnni3, type 2 ryanodine receptor (RyR2), Myh6, Myh7, and the Myh6/Myh7 ratio in murine iPSCWT and iPSCGSNOR-/-. (D) Representative immunofluorescence staining of α-actinin in outgrowths of beating iPSCWT and iPSCGSNOR-/- EBs on Day 10. Data are the mean ± SEM of 3 independent experiments. P-values were calculated using Student’s t-test. *P < 0.05.
The absence of GSNOR impacts Calcium handling
Ca2+ signaling within early cardiac progenitors may be important to promote sufficient differentiation for subsequent contractile function[33,34]. To obtain mechanistic insight into the onset of Ca2+ handling in our model and its impact on CM differentiation, recording of calcium transients with fluorescent indicators was performed. We compared Ca2+ handling in iPSCWT- and iPSCGSNOR-/--derived CMs on Day 10 and 21 of differentiation. Both strains exhibited properly shaped Ca2+ transients (spontaneous or electrically induced), Figure 7A and B, and a functional SR, since Ca2+ was released by activating the ryanodine receptor (RyR2) with 20 mM caffeine [Figure 7A]. However, iPSCGSNOR-/--CMs from Day 10 responded to 4 Hz pacing, whereas iPSCWT-CMs did not, although both strains responded to 4 Hz stimulation at Day 21 [Figure 7C]. Hence, we investigated the time dependence of changes in intracellular Ca2+ cycling in more detail. The Ca2+ transient peaks were higher in iPSCGSNOR-/- CMs (Figure 7C, P < 0.0001 on Day 10 and P < 0.0001 on Day 21) and the time to reach those Ca2+ transient peaks was shorter compared with iPSCWT CMs (Figure 7D, P < 0.0001 on Day 10 and P < 0.0016 on Day 21), suggesting a more efficient Ca2+-induced Ca2+ release (CICR) mechanism in cells lacking GSNOR. Likewise, the Ca2+ transient decay rate (shown as tt50%, Figure 7E) was, overall, significantly lower in iPSCGSNOR-/--CMs (Figure 7E, P < 0.0001 on Day 10). Next, we calculated the relative contribution of the three main mechanisms involved in the cytosolic Ca2+ uptake and removal (SERCA, NCX, Non-NCX) in our miPSC lines at Day 10 and 21 of differentiation [Figure 7F and G]. The main mechanism involved in Ca2+ decay in fully mature cells is re-uptake into the SR. Cytosolic Ca2+ re-uptake into the SR occurs through SERCA. NCX plays a major role in Ca2+ handling in early, developing CMs; however, it plays a more modest role in removing Ca2+ from the CM in mature cells. Consistently, SERCA activity (involved in SR Ca2+ re-uptake) was more prominent than the participation of Na+/Ca2+ exchanger (NCX) or other non-NCX mechanisms (both involved in removing Ca2+ from the cell) in both types of iPSCs. The contribution of SERCA at Day 10 was significantly higher in iPSCGSNOR-/- compared to iPSCWT (91.1% vs. 85.7%, respectively), depicting an advanced SR maturation in iPSCGSNOR-/-, whereas at Day 21 both iPSCs exhibited approximately 96% contribution by SERCA. Additionally, the contribution of NCX and Non-NCX was smaller in iPSCGSNOR-/- compared to iPSCWT on Day 10 and 21. Therefore, Ca2+ re-uptake into the SR was favored due to greater participation of SERCA over NCX in both groups, indicating an advanced state of SR maturation. However, intracellular Ca2+ was more efficiently handled in iPSCGSNOR-/- either as spontaneous Ca2+ transients or in response to electric field-stimulation. Overall, these data indicate that at Day 10 and 21 of differentiation, the iPSCGSNOR-/--CMs are more mature than iPSCWT-CMs.
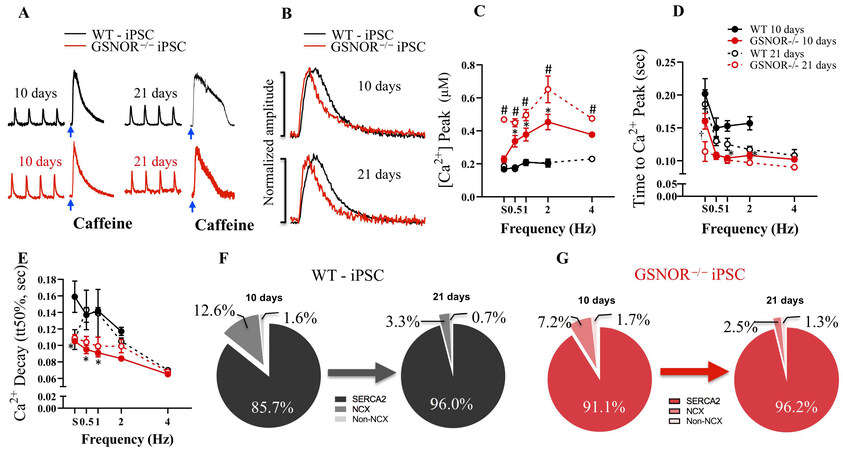
Figure 7. GSNOR loss of function promotes electrophysiologic maturation of miPSC-derived CMs. (A) Representative Ca2+ transients in iPSCWT (top, black) and iPSCGSNOR-/- (bottom, red) mice, under electric-field stimulation at 1 Hz and under caffeine-induced (τCaff, τCaff 0Na) at Day 10, and 21 during iPSC-CM differentiation. (B) Superimposed normalized Ca2+ transient amplitude from iPSCWT and iPSCGSNOR-/- mice at Day 10, and 21 after iPSC-CM differentiation. (C) Average Ca2+ peak in response to the increasing frequency of stimulation in iPSCWT (black) and iPSCGSNOR-/- (red) mice at Day 10 and 21 after iPSC-CM differentiation. (D) Average time to Ca2+ peak in response to the increasing frequency of stimulation in iPSCWT and iPSCGSNOR-/- mice at Day 10 and 21 after iPSC-CM differentiation. (E) Average time to half Ca2+ decay (tt50%) (from exponential fit) of iPSCWT (black) and iPSCGSNOR-/- (red) at Day 10 and 21 after iPSC-CM differentiation. (F) Relative contribution of the three Ca2+ transport systems (SERCA, NCX, non-NCX) in iPSCWT and (G) iPSCGSNOR-/- mice at Day 10 and 21 after iPSC-CM differentiation. Data are the mean ± SEM of 3 independent experiments. P-values were calculated using two-way ANOVA. (*P < 0.0001 on Day 10; #P < 0.0001 on Day 21; †P < 0.0016 on Day 21).
DISCUSSION
Our report is the first to show that GSNOR loss of function and S-nitrosylation more generally, accelerate the early stage of cardiomyogenesis as modeled by iPSCs. iPSC GSNOR-/- exhibited earlier onset EMT and increased cellular migration and proliferation. Moreover, the maturity of differentiating cardiomyocytes, as represented by the expression of structural proteins, Ca2+ transients and emergent genes involved in calcium handling, and sarcomere organization, revealed by α-actinin expression, occurred earlier in iPSCGSNOR-/-. GSNOR loss of function increased S-nitrosylation and decreased expression and function of GSK3β, with a concomitant increase of EMT-related transcription factors (Snail, Slug and Twist) but decreased E-cadherin [Figure 8], results that were also seen in iPSCWT and hiPSC following exposure to an NO donor. Nkx 2.5 was significantly increased upon directed differentiation toward cardiomyocytes, while brachyury, Isl1, and Gata4 mRNA were decreased in iPSCGSNOR-/- on Day 4. Therefore, GSNOR loss of function promoted the transition from mesoderm to cardiac progenitor and accelerated cardiomyocyte differentiation at the transcription level, as early as the start of adherent culture.
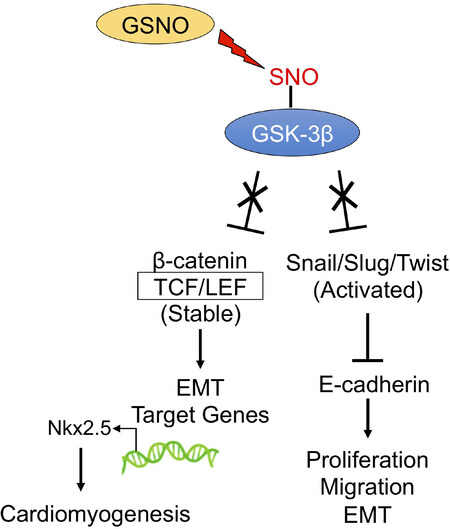
Figure 8. Schematic diagram of the proposed signaling mechanism that stimulates EMT and cardiomyogenesis. GSNOR loss of function results in GSNO accumulation and increased S-nitrosylation and reduced activity of GSK-3β, a protein that targets Snail/Slug/Twist and β-catenin for degradation through the ubiquitin proteosome pathway. This increased β-catenin and Snail/Slug/Twist represses E-cadherin, promotes β-catenin association with TCL/LEF complexes, augmenting transcription of proliferation, migration, EMT, and other target genes, resulting in elevated levels of Nkx2.5 mRNA levels and accelerating cardiomyogenesis.
Stem cell pluripotency and differentiation are regulated by several pathways. The differentiation and maintenance of pluripotency are dependent on NO concentration. Low NO concentrations delay stem cell differentiation, inducing the expression of Nanog, Oct4, and Sox2, and the activation of survival pathways[23], whereas high NO concentrations, as seen in iPSCGSNOR-/-, induce differentiation[35]. Consistent with this concept, we found that iPSCGSNOR-/- down-regulated Oct4 and Sox2; however, Klf4 was increased.
The major regulatory pathway of protein S-NO is via the metabolism of GSNO by GSNOR[36]. GSNO exists in a steady state with protein S-NO and cardiomyocytes use GSNO as a physiologic reservoir for NO, a major participant in trans-S-nitrosation reactions[37]. We showed that Gsnor mRNA and protein were present at Day 0 in iPSCWT but, as expected, not in iPSCGSNOR-/-, suggesting that the changes we observed are directly associated with the absence of GSNOR function.
At the start of adherent culture, on Day 4, cell proliferation/differentiation and the activation of EMT were seen only in iPSCGSNOR-/-. EMT is dependent on and orchestrated by Wnt/β-catenin signaling, consistent with its role during vertebrate heart development[38]. Wnt/β-catenin signaling may regulate the self-renewal of hPSCs[39]. Inactivation of GSK3β, a central regulator of the Wnt/β-catenin pathway, enables the accumulation of β-catenin, initially in the cytosol, and subsequently in the nucleus, where β-catenin forms a complex with transcription factor (TCF) proteins to activate Wnt pathway gene targets[40,41]. Wnt/β-catenin target genes regulate the expression of pluripotency and developmental factors associated with the primitive streak and early-stage germ layers[42]. Short-term Wnt induction maintains pluripotency, whereas long-term induction, in response to GSK3β inhibition, induces stem cell differentiation into endo- and mesoderm derivatives[43]. Increased S-nitrosylation of Gsk3β in iPSCGSNOR-/- and hiPSCs treated with GSNO likely maintains long-term Wnt induction, promoting expression of the EMT transcriptional activators, Snail, Slug, and Twist and reducing E-cadherin. However, the relationship between elevated NO and EMT is complicated. NO at subtoxic concentrations inhibits EMT and metastasis in metastatic cell lines by upregulating RKIP and E-cadherin protein[44]. These opposite effects may be related to an NO “threshold” that is dependent on cell type.
The regulation of Gsk3β activity is a complex process that can be mediated by multiple mechanisms including phosphorylation/dephosphorylation, S-nitrosylation, subcellular localization, protein-protein interactions, and proteolytic cleavage[45]. Subcellular localization of GSK3β can also modulate its enzymatic activity[15]. Moreover, S-nitrosylation of GSK3β inhibits its activity[15]. CM-specific deletion of GSK3 family members induces CM proliferation[46,47], and the degree of this proliferation is amplified in the setting of cardiac stress[48]. Our results also demonstrated that a reduction of GSK3β by S-nitrosylation resulted in increased proliferation but coupled with differentiation. However, the precise molecular mechanism for this shift from proliferating to terminally differentiated CMs is unclear, and currently of great scientific interest. Elucidating these mechanisms will provide novel strategies to reverse this terminally differentiated state, allowing CMs to resume proliferation.
Axins were significantly increased on Day 4, in iPSCGSNOR-/- cells. Both Axin and its homolog Axin2 act as scaffold proteins, which bind several components of the canonical Wnt signaling pathway[49]. Axin2, a bona fide downstream target of canonical Wnt, participates in a negative feedback loop, which could serve to limit the duration or intensity of a Wnt-initiated signal[50]. Moreover, Axin2 increases canonical TCF/LEF activity and Snail-mediated EMT progression[51]. Here, we observed increased levels of Axin2, which might indicate activation of the canonical Wnt pathway, likely secondary to the inactivation of GSK3β upon its S-nitrosylation. There may be at least two pools of GSK3β in cells, one associated with Axin and refractory to phosphorylation by Akt, and another that can be regulated by Akt[52]. Our findings complement a positive regulatory function for Axin1 and Axin2 in Wnt signaling[53] in developing CMs.
The differentiation of stem cells along the cardiac lineage is characterized by the early expression of a network of mesodermal-cardiac transcription factors, that activate gene expression for structural proteins such as MHC, Mlc2v, α-actinin, and troponin. Upon directed differentiation toward cardiomyocytes, pluripotent cells first pass through a progenitor stage, identified by the expression of specific transcription factors, such as brachyury in the mesoderm stage (early phase) and Nkx 2.5, Gata4, Mef2c in the cardiac progenitor cell stage (later phase)[54]. We showed that iPSCsGSNOR-⁄- expression of markers of mesoderm induction (t-brachyury); cardiac mesoderm (Isl1); and cardiac progenitors (Gata4) were reduced, whereas, Nkx 2.5, a cardiac progenitor marker, was increased. In murine embryonic stem cells, exposure to the NO donor, diethylenetriamine/nitric oxide adduct can repress brachyury, Gata4, Gata6, Fibroblast growth factor (Fgf) 5, and Fgf8[23]. Following cardiogenic differentiation, Isl1 is progressively repressed, and only a small number of Isl1+ cells remain in the postnatal heart[55].
Similar to other post-translational modifications (i.e., phosphorylation, methylation, acetylation), S-NO regulates the expression and function of key proteins controlling mammalian cell differentiation[5] and cell-cycle activity[56]. In the adult heart, S-NO modulates signaling pathways important for vasodilation, cardiomyocyte contraction, mitochondrial function[57], and Ca2+ handling[3,58]. In murine embryonic stem cells[2], an isotope labeling proteomics screening approach, demonstrated that Prdx-2 nitrosylation (following GSNO exposure) favors cardiomyocyte differentiation. Although an increased percentage of beating cells was observed in iPSCsGSNOR-/- on Day 8, significant changes occur as the cells continue to develop. Therefore, we followed the progress of cell differentiation until Day 21. In addition, NO improves calcium handling in mature cardiomyocytes[59]; however, the acute and chronic effects on differentiating cardiomyocytes have not been elucidated.
There is no current understanding regarding the onset of Ca2+ handling and its impact on differentiation and cardiogenesis. However, Ca2+ transients, calcium handling-related gene, protein expression, and sarcomere organization were, except for Tnnt2, all significantly increased on Days 10 and 21 in iPSCGSNOR-/- compared to iPSCWT. These results are supported by the observation that the NO-donor, CysNO, upregulated βMHC, producing a higher βMHC to αMHC ratio and superior calcium handling capabilities in EBs without altering EB size or viability[1]. Our findings also corroborate that RyR2 S-nitrosylation increases the activity of this channel[60]; whereas, decreased RyR2 S-nitrosylation is associated with reduced SR Ca2+ release[61].
Prolonging time in culture promotes the maturation of iPSC-CMs, including more mature morphology, structure, and physiology[62]. Our iPSC-derived CMs were grown for only 21 days. Human iPSC-derived CMs chronologically aged in vitro, exhibited accelerated senescence and functional deterioration within four months in culture, resembling cardiac tissue from a 65-year-old human[63]. During senescence and mammalian aging, GSNOR expression declines, resulting in nitrosative stress and the accumulation of damaged mitochondria[64]. In contrast, centenarians maintain high GSNOR expression[64].
This study is the first to investigate the effects of GSNOR deficiency during cardiomyocyte differentiation as modeled by iPSC. However, there are limitations to this study. First, this study is limited to the early stages of differentiation of both murine and human iPSCs. Second, the heterogeneous composition of differentiating iPSCs could mask the magnitude of gene expression changes. Further evaluation, such as a directed differentiation with a monolayer culture, will allow for dissection of different stages of cardiac differentiation and a better understanding of the role of GSNOR at different stages of cardiomyocyte differentiation.
In conclusion, the absence of GSNOR accelerates cardiomyocyte maturation from iPSCs. We observed increased S-nitrosylation of GSK3β, cellular migration, proliferation, and induction of EMT. Together, these findings have important implications and may offer insights into improving cell therapy and enhancing disease modeling in the cardiovascular system.
DECLARATIONS
Authors’ contributionsDesigned the study and performed experiments: Salerno AG, Wanschel ACBA, Dulce RA, Hatzistergos KE
Prepared the manuscript: Salerno AG, Wanschel ACBA, Dulce RA, Hatzistergos KE, Balkan W, Hare JM
Designed experiments, guided the interpretation of the results and edited the manuscript: Balkan W,
Not applicable.
Financial support and sponsorshipThis work was supported by the National Institutes of Health grants R01 HL107110, R01 HL137355, R01 HL084275 and 5UM HL113460; grants from the Starr Foundation and the Soffer Family Foundation (all awarded to Hare JM); and a São Paulo Research Foundation (FAPESP) grant# 2016/01044-0 (Salerno AG) and grant# 2016/01746-4 (Wanschel ACBA).
Conflicts of interestHare JM reported having a patent for cardiac cell-based therapy. He holds equity in Vestion Inc. and maintains a professional relationship with Vestion Inc. as a consultant and member of the Board of Directors and Scientific Advisory Board. Hare JM is the Chief Scientific Officer, a compensated consultant and advisory board member for Longeveron, and holds equity in Longeveron. Hare JM is also the co-inventor of intellectual property licensed to Longeveron. Longeveron LLC and Vestion Inc. did not participate in funding this work. Dr. Hare’s relationships are disclosed to the University of Miami, and a management plan is in place. Other authors declared that there are no conflicts of interest.
Ethical approval and consent to participateThe Institutional Animal Care and Use Committee (IACUC) approved the animal studies described in this paper as Protocol #20-168.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Hodge AJ, Zhong J, Lipke EA. Enhanced stem cell-derived cardiomyocyte differentiation in suspension culture by delivery of nitric oxide using S-nitrosocysteine. Biotechnol Bioeng 2016;113:882-94.
2. Wu B, Yu H, Wang Y, et al. Peroxiredoxin-2 nitrosylation facilitates cardiomyogenesis of mouse embryonic stem cells via XBP-1s/PI3K pathway. Free Radic Biol Med 2016;97:179-91.
3. Dulce RA, Mayo V, Rangel EB, Balkan W, Hare JM. Interaction between neuronal nitric oxide synthase signaling and temperature influences sarcoplasmic reticulum calcium leak: role of nitroso-redox balance. Circ Res 2015;116:46-55.
4. Haldar SM, Stamler JS. S-nitrosylation: integrator of cardiovascular performance and oxygen delivery. J Clin Invest 2013;123:101-10.
5. Cao Y, Gomes SA, Rangel EB, et al. S-nitrosoglutathione reductase-dependent PPARγ denitrosylation participates in MSC-derived adipogenesis and osteogenesis. J Clin Invest 2015;125:1679-91.
6. Beigi F, Gonzalez DR, Minhas KM, et al. Dynamic denitrosylation via S-nitrosoglutathione reductase regulates cardiovascular function. Proc Natl Acad Sci U S A 2012;109:4314-9.
7. Chung HS, Murray CI, Venkatraman V, et al. Dual labeling biotin switch assay to reduce bias derived from different cysteine subpopulations: a method to maximize S-nitrosylation detection. Circ Res 2015;117:846-57.
8. Liu L, Yan Y, Zeng M, et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 2004;116:617-28.
9. Hatzistergos KE, Paulino EC, Dulce RA, et al. S-nitrosoglutathione reductase deficiency enhances the proliferative expansion of adult heart progenitors and myocytes post myocardial infarction. J Am Heart Assoc 2015;4:e001974.
10. Ohta S, Suzuki K, Tachibana K, Tanaka H, Yamada G. Cessation of gastrulation is mediated by suppression of epithelial-mesenchymal transition at the ventral ectodermal ridge. Development 2007;134:4315-24.
11. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 2006;7:131-42.
12. Zhou BP, Deng J, Xia W, et al. Dual regulation of snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol 2004;6:931-40.
13. Burridge PW, Matsa E, Shukla P, et al. Chemically defined generation of human cardiomyocytes. Nat Methods 2014;11:855-60.
14. Kattman SJ, Witty AD, Gagliardi M, et al. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 2011;8:228-40.
15. Wang SB, Venkatraman V, Crowgey EL, et al. Protein S-nitrosylation controls glycogen synthase Kinase 3β function independent of its phosphorylation state. Circ Res 2018;122:1517-31.
16. Uosaki H, Magadum A, Seo K, et al. Identification of chemicals inducing cardiomyocyte proliferation in developmental stage-specific manner with pluripotent stem cells. Circ Cardiovasc Genet 2013;6:624-33.
18. Lian X, Hsiao C, Wilson G, et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A 2012;109:E1848-57.
19. Forrester MT, Thompson JW, Foster MW, Nogueira L, Moseley MA, Stamler JS. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol 2009;27:557-9.
20. Bloch W, Fleischmann BK, Lorke DE, et al. Nitric oxide synthase expression and role during cardiomyogenesis. Cardiovasc Res 1999;3:675-84.
21. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861-72.
22. Li HX, Han M, Bernier M, et al. Krüppel-like factor 4 promotes differentiation by transforming growth factor-beta receptor-mediated Smad and p38 MAPK signaling in vascular smooth muscle cells. J Biol Chem 2010;285:17846-56.
23. Tejedo JR, Tapia-Limonchi R, Mora-Castilla S, et al. Low concentrations of nitric oxide delay the differentiation of embryonic stem cells and promote their survival. Cell Death Dis 2010;1:e80.
24. Saraiva RM, Minhas KM, Raju SV, et al. Deficiency of neuronal nitric oxide synthase increases mortality and cardiac remodeling after myocardial infarction: role of nitroso-redox equilibrium. Circulation 2005;112:3415-22.
25. Fu K, Chronis C, Soufi A, et al. Comparison of reprogramming factor targets reveals both species-specific and conserved mechanisms in early iPSC reprogramming. BMC Genomics 2018;19:956.
26. Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J Cell Biol 2005;168:29-33.
27. Wang Y, Liu J, Ying X, Lin PC, Zhou BP. Twist-mediated epithelial-mesenchymal transition promotes breast tumor cell invasion via inhibition of hippo pathway. Sci Rep 2016;6:24606.
28. Otani R, Mukasa A, Shin M, et al. Brachyury gene copy number gain and activation of the PI3K/Akt pathway: association with upregulation of oncogenic Brachyury expression in skull base chordoma. J Neurosurg 2018;128:1428-37.
29. Yan X, Li Z, Li H, et al. Inhibition of glycogen synthase kinase 3 beta suppresses the growth and survival of skull base chordoma cells by downregulating brachyury expression. Onco Targets Ther 2019;12:9783-91.
30. Zhu J, Kwan KM, Mackem S. Putative oncogene brachyury (T) is essential to specify cell fate but dispensable for notochord progenitor proliferation and EMT. Proc Natl Acad Sci U S A 2016;113:3820-5.
31. Bedada FB, Chan SS, Metzger SK, et al. Acquisition of a quantitative, stoichiometrically conserved ratiometric marker of maturation status in stem cell-derived cardiac myocytes. Stem Cell Reports 2014;3:594-605.
32. Weber N, Schwanke K, Greten S, et al. Stiff matrix induces switch to pure β-cardiac myosin heavy chain expression in human ESC-derived cardiomyocytes. Basic Res Cardiol 2016;111:68.
33. Mesaeli N, Nakamura K, Zvaritch E, et al. Calreticulin is essential for cardiac development. J Cell Biol 1999;144:857-68.
34. Li J, Pucéat M, Perez-Terzic C, et al. Calreticulin reveals a critical Ca(2+) checkpoint in cardiac myofibrillogenesis. J Cell Biol 2002;158:103-13.
35. Mora-Castilla S, Tejedo JR, Hmadcha A, et al. Nitric oxide repression of Nanog promotes mouse embryonic stem cell differentiation. Cell Death Differ 2010;17:1025-33.
36. Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol 2009;10:721-32.
37. Kohr MJ, Aponte AM, Sun J, et al. Characterization of potential S-nitrosylation sites in the myocardium. Am J Physiol Heart Circ Physiol 2011;300:H1327-35.
38. Lickert H, Kutsch S, Kanzler B, Tamai Y, Taketo MM, Kemler R. Formation of multiple hearts in mice following deletion of β-catenin in the embryonic endoderm. Dev Cell 2002;3:171-81.
39. Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med 2004;10:55-63.
40. McCubrey JA, Steelman LS, Bertrand FE, et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014;5:2881-911.
41. Nakamura M, Liu T, Husain S, et al. Glycogen Synthase kinase-3α promotes fatty acid uptake and lipotoxic cardiomyopathy. Cell Metab 2019;29:1119-1134.e12.
42. Hödar C, Assar R, Colombres M, et al. Genome-wide identification of new Wnt/beta-catenin target genes in the human genome using CART method. BMC Genomics 2010;11:348.
43. Huang TS, Li L, Moalim-Nour L, et al. A regulatory network involving β-catenin, e-cadherin, PI3k/Akt, and slug balances self-renewal and differentiation of human pluripotent stem cells in response to wnt signaling. Stem Cells 2015;33:1419-33.
44. Baritaki S, Huerta-Yepez S, Sahakyan A, et al. Mechanisms of nitric oxide-mediated inhibition of EMT in cancer: inhibition of the metastasis-inducer Snail and induction of the metastasis-suppressor RKIP. Cell Cycle 2010;9:4931-40.
45. Mirzoev TM, Sharlo KA, Shenkman BS. The role of GSK-3β in the regulation of protein turnover, myosin phenotype, and oxidative capacity in skeletal muscle under disuse conditions. Int J Mol Sci 2021;22:5081.
46. Woulfe KC, Gao E, Lal H, et al. Glycogen synthase kinase-3beta regulates post-myocardial infarction remodeling and stress-induced cardiomyocyte proliferation in vivo. Circ Res 2010;106:1635-45.
47. Matsuda T, Zhai P, Maejima Y, et al. Distinct roles of GSK-3alpha and GSK-3beta phosphorylation in the heart under pressure overload. Proc Natl Acad Sci U S A 2008;105:20900-5.
48. Singh AP, Umbarkar P, Guo Y, Force T, Gupte M, Lal H. Inhibition of GSK-3 to induce cardiomyocyte proliferation: a recipe for in situ cardiac regeneration. Cardiovasc Res 2019;115:20-30.
49. Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol 2002;22:1172-83.
50. Kang HE, Seo Y, Yun JS, et al. Metformin and niclosamide synergistically suppress Wnt and YAP in APC-mutated colorectal cancer. Cancers (Basel) 2021;13:3437.
51. Ahn SY, Kim NH, Lee K, et al. Niclosamide is a potential therapeutic for familial adenomatosis polyposis by disrupting Axin-GSK3 interaction. Oncotarget 2017;8:31842-55.
52. Ding VW, Chen RH, McCormick F. Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J Biol Chem 2000;275:32475-81.
53. Lui TT, Lacroix C, Ahmed SM, et al. The ubiquitin-specific protease USP34 regulates axin stability and Wnt/β-catenin signaling. Mol Cell Biol 2011;31:2053-65.
54. Kehat I, Kenyagin-Karsenti D, Snir M, et al. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest 2001;108:407-14.
55. Weinberger F, Mehrkens D, Friedrich FW, et al. Localization of Islet-1-positive cells in the healthy and infarcted adult murine heart. Circ Res 2012;110:1303-10.
56. Kunieda T, Minamino T, Miura K, et al. Reduced nitric oxide causes age-associated impairment of circadian rhythmicity. Circ Res 2008;102:607-14.
57. Murray CI, Kane LA, Uhrigshardt H, Wang SB, Van Eyk JE. Site-mapping of in vitro S-nitrosation in cardiac mitochondria: implications for cardioprotection. Mol Cell Proteomics 2011;10:M110.004721.
58. Gonzalez DR, Treuer A, Sun QA, Stamler JS, Hare JM. S-nitrosylation of cardiac ion channels. J Cardiovasc Pharmacol 2009;54:188-95.
59. Petroff MG, Kim SH, Pepe S, et al. Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+ release in cardiomyocytes. Nat Cell Biol 2001;3:867-73.
60. Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 1998;279:234-7.
61. Gonzalez DR, Beigi F, Treuer AV, Hare JM. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci U S A 2007;104:20612-7.
62. Ahmed RE, Anzai T, Chanthra N, Uosaki H. A brief review of current maturation methods for human induced pluripotent stem cells-derived cardiomyocytes. Front Cell Dev Biol 2020;8:178.
63. Acun A, Nguyen TD, Zorlutuna P. In vitro aged, hiPSC-origin engineered heart tissue models with age-dependent functional deterioration to study myocardial infarction. Acta Biomater 2019;94:372-91.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Salerno AG, Wanschel ACBA, Dulce RA, Hatzistergos KE, Balkan W, Hare JM. S-nitrosoglutathione reductase (GSNOR) deficiency accelerates cardiomyocyte differentiation of induced pluripotent stem cells. J Cardiovasc Aging 2021;1:13. http://dx.doi.org/10.20517/jca.2021.19
AMA Style
Salerno AG, Wanschel ACBA, Dulce RA, Hatzistergos KE, Balkan W, Hare JM. S-nitrosoglutathione reductase (GSNOR) deficiency accelerates cardiomyocyte differentiation of induced pluripotent stem cells. The Journal of Cardiovascular Aging. 2021; 1(1): 13. http://dx.doi.org/10.20517/jca.2021.19
Chicago/Turabian Style
Salerno, Alessandro G., Amarylis C. B. A. Wanschel, Raul A. Dulce, Konstantinos E. Hatzistergos, Wayne Balkan, Joshua M. Hare. 2021. "S-nitrosoglutathione reductase (GSNOR) deficiency accelerates cardiomyocyte differentiation of induced pluripotent stem cells" The Journal of Cardiovascular Aging. 1, no.1: 13. http://dx.doi.org/10.20517/jca.2021.19
ACS Style
Salerno, AG.; Wanschel ACBA.; Dulce RA.; Hatzistergos KE.; Balkan W.; Hare JM. S-nitrosoglutathione reductase (GSNOR) deficiency accelerates cardiomyocyte differentiation of induced pluripotent stem cells. J. Cardiovasc. Aging. 2021, 1, 13. http://dx.doi.org/10.20517/jca.2021.19
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 13 clicks
Cite This Article 13 clicks

 Like This Article 18
likes
Like This Article 18
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.