
Hematopoietic JAK2V617F-mediated clonal hematopoiesis: AIM2 understand mechanisms of atherogenesis


The age-dependent accumulation of somatic DNA mutations in “driver” genes within the hematopoietic system can provide a fitness advantage to the mutant cells and thus allow for their aberrant clonal expansion, a process known as clonal hematopoiesis (CH). While this process is clearly recognized to initiate cancers in the hematopoietic system, numerous studies have documented that this condition is remarkably widespread in the general population and that individuals who harbor these mutations rarely go on to develop a hematologic malignancy. Thus, this condition has been referred to as clonal hematopoiesis of indeterminate potential or age-related clonal hematopoiesis. Multiple lines of evidence indicate that this prevalent form of CH is associated with higher rates of mortality, and cause-specific analyses indicate that CH resulting from mutations in any one of a number of driver genes will increase the incidence of coronary heart disease and ischemic stroke. Individuals with clonal hematopoiesis due to the JAK2V617Fdriver mutation are reported to have a 12-times higher risk for myocardial infarction, while other major CH driver gene mutations (e.g., TET2, DNMT3A, ASXL1) increased the risk by 1.7-2.0-fold[1]. Additionally, it has been reported that JAK2V617F-mediated CH increases the risk of developing venous thrombosis[2]. Therefore, individuals with the JAK2V617F mutation in hematopoietic cells may be at a particularly high risk of developing cardiovascular disease (CVD).
The JAK2V617Fmutation, which leads to the constitutive activation of JAK2-associated cytokine receptor signaling, was originally recognized as being frequently mutated in myeloproliferative neoplasms (MPN) such as polycythemia vera and essential thrombocytosis. However, it has been reported that the JAK2V617F mutation can be detected at a low allele fraction in as many as 3.1% of the general population using a highly sensitive ddPCR methodology[3]. Thus, the apparent widespread detection of JAK2V617F-mediated clonal hematopoiesis has led to interest in experimental studies to elucidate the mechanisms by which the JAK2V617F mutation in hematopoietic cells can contribute to CVD.
In a previous study, this group reported that pan-hematopoietic Jak2V617Fmutation (the Jak2 mutation in mice is relevant to human JAK2V617F) promotes atherogenesis in hyperlipidemic mice[4]. Specifically, the analysis of plaque composition revealed that the hematopoietic Jak2 mutation increases neutrophil infiltration in early atherosclerotic lesions and a larger necrotic core in the more advanced lesion, consistent with enhanced rolling and adhesion capacity of Jak2-mutant neutrophils (see Figure 1). Increased erythrophagocytosis and decreased efferocytosis by macrophages within the lesion were also observed in mice with the Jak2V617F mutation. In addition, red blood cells, either mouse or human, were more prone to erythrophagocytosis, presumably because of decreased expression of “don’t-eat-me” signals. Collectively, these results highlighted numerous possible mechanisms through which JAK2V617F-mediated CH could increase the risk of atherosclerotic CVD. However, a significant limitation of this study is that the pan-hematopoietic Jak2V617F mouse model displays MPN phenotypes, including increased hematocrit or platelet counts, that are not observed in individuals with CH. Thus, more refined models of JAK2V617F-mediated CH are required to more rigorously understand the nature of the CVD mechanisms.
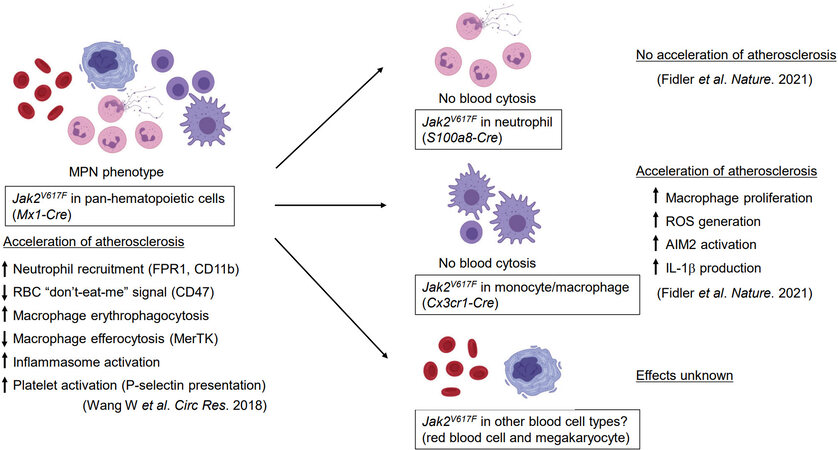
Figure 1. Experimental studies of the Jak2V617F mutation in blood and its effects on atherogenesis.
The creation of a model of JAK2-CH is not straightforward because multiple mouse lines that harbor the Jak2V617F mutation in their hematopoietic stem and progenitor cells (HSPC) exhibit MPN phenotypes that confound the CVD phenotype under investigation. Thus, a strategy to the model of JAK2-CH is to establish mice that express the mutant JAK2/Jak2 gene in specific blood cell populations such as neutrophils or monocytes/macrophages and potentially avoid confounding MPN phenotypes. In this regard, Sano et al.[5] used a lentivirus vector encoding human JAK2V617F under myeloid-specific synthetic SP146 promoter/gp91 enhancer combination. This system avoids using the Lyz2-Cre mice to express the mutant transgene because this system is “leaky” and leads to low JAK2V617Fexpression in HSPC and the MPN-associated expansion of mutant blood cells. In contrast, bone marrow reconstruction of lethally irradiated mice with HSPC transduced with the lentivirus construct allowed JAK2V617Fto be expressed only in myeloid cell lineages, and this avoids the MPN-like hematological abnormalities. These myeloid-specific JAK2V617F mice exhibited accelerated cardiac dysfunction following experimental myocardial infarction or pressure overload-mediated hypertrophy. Moreover, these heart failure phenotypes were associated with cytokine activation, including IL-1β, in the myocardium.
In the more recent study, Fidler et al.[6] utilized Cre/LoxP-mediated recombination to achieve Jak2V617F expression in specific myeloid cell types. Specifically, S100a8-Cre and Cx3cr1-Cre mice were employed to achieve neutrophil- and monocyte/macrophage-specific Jak2V617F expression, respectively (see Figure 1). Similar to the earlier findings[5], neither of these strains displayed MPN characteristics, thereby allowing the analysis of CVD phenotypes of Jak2V617F in each myeloid population in the absence of elevated blood cell counts. It was found that, hyperlipidemic mice reconstituted with bone marrow cells from Cx3cr1-Cre/Jak2V617F mice, exhibited increased atherosclerotic plaque size and signs of vulnerable plaque despite lower serum low-density-lipoprotein cholesterol and triglyceride levels[6]. Consistent with larger necrotic core, plaques in the Cx3cr1-Cre/Jak2V617F mice also contained greater numbers of macrophages due to a higher proliferation rate. On the other hand, mice with bone marrow cells from S100a8-Cre/Jak2V617F mice did not display increased atherosclerosis progression. This finding is somewhat unexpected, considering the previous report that Jak2V617Fincreases neutrophil rolling and adhesion to vascular wall[4] and the current understanding that neutrophils are associated with atherogenesis[7]. Regardless, these analyses highlight the importance of rigorous cell type-specific expression analyses to delineate the primary cell types that mediate the atherogenic effects of CH.
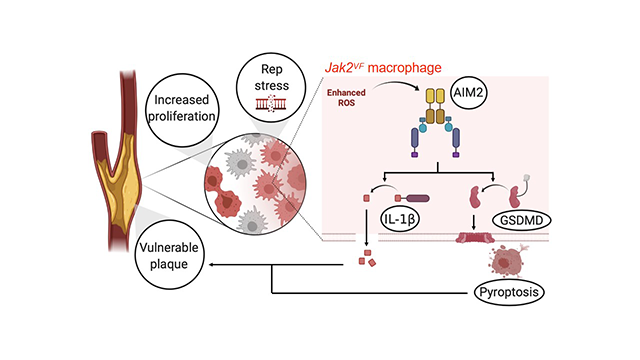
Fidler et al.[6] reported that Jak2V617F macrophages express higher levels of the AIM2 inflammasome. Similarly, increased AIM2 expression has been observed in JAK2V617F-expressing human monocyte/macrophage THP-1 cells[5]. Further analyses revealed that AIM2 is regulated by elevated IFNg signaling which, in turn, is activated by increased oxidative DNA damage and replication stress[6]. The activation of AIM2 leads to the over-production of IL-1β and IL-18 and also promotes Gasdermin-mediated pyroptosis. Notably, it was shown that deficiency of the AIM2 inflammasome or inhibition of IL-1β signaling largely reversed the atherosclerosis phenotype driven by Jak2V617F in hematopoietic cells, whereas deficiency of the NLRP3 inflammasome did not show protection from atherosclerosis under these conditions. In contrast, pharmacological inhibition of the NLRP3 inflammasome inhibited atherogenesis and heart failure in models of mutant Tet2-mediated clonal hematopoiesis[8,9]. Collectively, these data raise the exciting possibility of mutant gene-specific differences in how CH modulates CVD. Thus, by extension of these findings, there may be a rationale for personalized therapies based upon the diagnosis of the specific gene(s) mutated in CH.
JAK2V617F-mutant HSPC can give rise to multiple lineages of blood cells including myeloid cells, platelets, red blood cells, etc. Thus, the development of CVD models with selective Jak2V617F expression in additional blood cell lineages is warranted. A previous study has shown that Jak2V617F-mutant neutrophils display increased neutrophil extracellular trap (NET) formation in both humans and mice, which is associated with venous thrombosis formation[2]. As discussed, Jak2V617F-mutant macrophages, but not neutrophils, are contribute to enhanced atherogenesis through an AIM2 inflammasome-dependent mechanism[7]. Given these findings, it would be of interest to assess whether JAK2-CH is enriched in patients with plaque erosion, as this form of CVD event is associated with NET formation. Finally, it would also be of interest to examine the pathogenic roles of JAK2V617F in other blood cell types using selective expression strategies in mice.
DECLARATIONS
Authors’ contributionsDrafted and revised the manuscript: Sano S, Walsh K
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was funded by National Institutes of Health grants HL131006, HL132564, HL139819, HL141256, HL138014 and AG072095 to Walsh K and HL152174 to Sano S and Walsh K.
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 2017;377:111-21.
2. Wolach O, Sellar RS, Martinod K, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med 2018;10:eaan8292.
3. Cordua S, Kjaer L, Skov V, et al. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood 2019;134:469-79.
4. Wang W, Liu W, Fidler T, et al. Macrophage inflammation, erythrophagocytosis, and accelerated atherosclerosis in Jak2 (V617F) mice. Circ Res 2018;123:e35-47.
5. Sano S, Wang Y, Yura Y, et al. JAK2 (V617F)-mediated clonal hematopoiesis accelerates pathological remodeling in murine heart failure. JACC Basic Transl Sci 2019;4:684-97.
6. Fidler TP, Xue C, Yalcinkaya M, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 2021;592:296-301.
7. Silvestre-Roig C, Braster Q, Wichapong K, et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019;569:236-40.
8. Fuster JJ, MacLauchlan S, Zuriaga MA, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017;355:842-7.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Sano S, Walsh K. Hematopoietic JAK2V617F-mediated clonal hematopoiesis: AIM2 understand mechanisms of atherogenesis. J Cardiovasc Aging 2021;1:5. http://dx.doi.org/10.20517/jca.2021.06
AMA Style
Sano S, Walsh K. Hematopoietic JAK2V617F-mediated clonal hematopoiesis: AIM2 understand mechanisms of atherogenesis. The Journal of Cardiovascular Aging. 2021; 1(1): 5. http://dx.doi.org/10.20517/jca.2021.06
Chicago/Turabian Style
Sano, Soichi, Kenneth Walsh. 2021. "Hematopoietic JAK2V617F-mediated clonal hematopoiesis: AIM2 understand mechanisms of atherogenesis" The Journal of Cardiovascular Aging. 1, no.1: 5. http://dx.doi.org/10.20517/jca.2021.06
ACS Style
Sano, S.; Walsh K. Hematopoietic JAK2V617F-mediated clonal hematopoiesis: AIM2 understand mechanisms of atherogenesis. J. Cardiovasc. Aging. 2021, 1, 5. http://dx.doi.org/10.20517/jca.2021.06
About This Article
Copyright

Data & Comments
Data


 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 39
likes
Like This Article 39
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.