
Immune mechanisms of cardiac aging

 ,
, Abstract
Advances in healthcare and improvements in living conditions have led to rising life expectancy worldwide. Aging is associated with excessive oxidative stress, a chronic inflammatory state, and limited tissue healing, all of which result in an increased risk of heart failure. In fact, the prevalence of heart failure approaches 40% in the ninth decade of life, with the majority of these cases suffering from heart failure with preserved ejection fraction (HFpEF). In cardiomyocytes (CMs), age-related mitochondrial dysfunction results in disrupted calcium signaling and covalent protein-linked aggregates, which cause cardiomyocyte functional disturbances, resulting in increased stiffness and diastolic dysfunction. Importantly, aging is also associated with chronic low-grade, sterile inflammation, which alters the function of interstitial cardiac cells and leads to cardiac fibrosis. Taken together, cardiac aging is associated with cellular, structural, and functional changes in the heart that contribute to the rising prevalence of heart failure in older people.
Keywords
INTRODUCTION
The percentage of people above the age of 65 increased to 16.9% in 2020, and this figure is expected to reach 22% by 2050. As the population ages, there is an increasing focus on understanding cardiac pathophysiological changes associated with aging. These changes include an increased risk of cardiovascular disease, heart failure, and changes in the structure and function of the heart. Additionally, the aging process can lead to changes in the heart’s electrical conduction system, leading to arrhythmias. As such, there is a need for further research into the mechanisms underlying these changes and potential interventions to reduce the risk of cardiovascular disease in older people. Aging is associated with a chronic low-grade inflammation, termed inflammaging, which describes the normal aging process and the associated increased risk of age-related diseases. It is characterized by a persistent low-grade inflammatory response that can be caused by a variety of factors, including cellular senescence, immune system dysfunction, and increased organ stress. The effects of inflammaging on the heart are far-reaching and may include an increased risk of cardiovascular diseases, including coronary artery disease, stroke, and heart failure. This review discusses major pathophysiological pathways that lead to cardiac aging, focusing on the role of inflammaging.
CARDIAC FUNCTIONAL AND STRUCTURAL CHANGES DURING AGING
Aging is associated with a constellation of left ventricular (LV) hypertrophy, left atrial enlargement, and interstitial fibrosis with diastolic dysfunction, with no changes in systolic function. These structural changes are characteristic of HFpEF and are typically associated with a significant reduction in exercise capacity. Age-specific cardiac imaging studies documented an increased prevalence of diastolic dysfunction with aging. Cardiac magnetic resonance (CMR) studies documented a significant 5 mg/mL/year increase in LV mass/volume ratio with a corresponding 0.4 mL/year reduction in stroke volume owing to the thickened cardiac muscle dysregulation[1]. Echocardiographic studies reached similar conclusions and demonstrated that the prevalence of diastolic dysfunction increased exponentially with age[1]. While echocardiographic parameters of diastolic dysfunction were rarely observed in individuals younger than 50, over half of the individuals between 70 and 80 years of age had echocardiographic evidence of diastolic dysfunction. This prevalence rises to two-thirds of study participants older than 80 years of age[2].
Cardiac aging is also associated with electrophysiological changes that result in decreased intrinsic heart rate, response to adrenergic signaling, and increased arrhythmogenic effects[3]. Combined with diastolic dysfunction seen in aging, which reduces ventricular compliance and stroke volume, reduced intrinsic heart rate with aging results in diminished cardiac output. Furthermore, alterations to adrenergic and cholinergic signaling within CMs result in their hypertrophy as well as cardiac fibrosis, contributing to age-related structural and functional changes[4,5]. Combined with the limited ability of the heart muscle to regenerate, excessive cellular loss contributes to cardiac dysfunction in aging. Therefore, structural changes in the heart with aging have been linked to the development of heart failure, particularly with preserved ejection fraction.
In addition to changes in the ventricles, aging is associated with important structural and functional changes affecting the atria. Aging-associated oxidative stress, inflammation, cellular senescence, and extracellular matrix remodeling increase atrial fibrosis resulting in a decrease in contractility and conduction velocity[6,7]. Atrial fibrosis can then impair atrial electrical conduction and mechanical function, creating a substrate for atrial fibrillation (AF) initiation and maintenance and the associated risk of embolic stroke.
CELLULAR PATHWAYS ASSOCIATED WITH INFLAMMAGING
Aging is associated with a chronic low-grade sterile inflammatory state, termed “inflammaging”, which is one of the hallmark features of aging and can be linked to increased morbidity and mortality in older individuals[8]. Sterile inflammation is a type of inflammation that is triggered by non-infectious stimuli, such as increased cellular senescence, immunosenescence, dysregulated inflammasome activation, mitochondrial dysfunction, age-related autophagy and mitophagy defects[9], and systemic predisposing factors such as changes in the number and composition of intestinal microbiota[10,11] and clonal hematopoiesis of indeterminate potential (CHIP)[12]. Sterile inflammation involves the activation of innate immune cells, such as macrophages and neutrophils, by damage-associated molecular patterns (DAMPs) that bind to pattern recognition receptors (PRRs) on immune cells. Sterile inflammation can have beneficial effects, such as promoting wound healing and tissue repair, but it can also have harmful effects, such as causing chronic diseases, organ damage, or autoimmunity. Consequently, multiple studies have documented increased levels of inflammatory cytokines in the peripheral blood of healthy older individuals, even in the absence of underlying stress or infection. The risk and consequences of this inflammatory response affect the heart. There is evidence of increased activation of inflammatory pathways, e.g., increased levels of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which could lead to the production of pro-inflammatory cytokines, and pro-inflammatory macrophages in older hearts, particularly in females[13-15].
Mechanistically, mitochondrial dysfunction plays a central role in the aging process and is at the crossroads of central pathways involved in inflammaging[16-18]. Aging is associated with mitochondrial structural abnormalities resulting in functional deterioration and increased reactive oxygen species (ROS) generation. In addition, aging is associated with reduced activation of 5' AMP-activated protein kinase (AMPK) and sirtuins (SIRT), among other key regulators of mitochondrial function, cellular energy balance, and homeostasis[19]. Indeed, animal models of cardiac hypertrophy documented the link between a reduction in SIRT6 expression and the development of hypertrophy and diastolic dysfunction[20]. Additionally, the expression of SIRT3, an essential regulator of mitochondrial function, is reduced with aging, resulting in endothelial cell dysfunction and diastolic dysfunction[21]. Furthermore, the downregulation of AMPK and sirtuins results in a shift of mitochondrial oxidation favoring glycolytic pathways, which promotes inflammation in multiple cell lines, including coronary endothelial cells[22,23]. In addition to the effects of AMPK and sirtuins on the mitochondria and energy homeostasis, these pathways play a critical role in modulating NF-κB activation, and the downregulation of both these pathways results in uninhibited NF-κB activity and exacerbates inflammatory responses[15,24]. Mitochondrial dysfunction and disturbances in cellular homeostasis result in the release of mitochondrial DNA (mtDNA), eventually leading to increased inflammation through the activation of toll-like receptors (TLRs) and the nucleotide-binding oligomerization domain-3 (NLRP3) inflammasome. Beyond the role of mitochondrial dysfunction in inflammation, mitochondrial dysfunction leads to disturbed calcium signaling due to changes in the type 2 ryanodine receptor (RyR2) and the sarcoplasmic reticulum Ca2+ ATPase pump (SERCA), thus contributing to cardiomyocyte dysfunction and diastolic heart failure.
Aging is associated with a plethora of pathophysiological changes with a resulting chronic low-grade inflammatory state [Figure 1]. Cellular senescence, a phenomenon exacerbated by aging, is caused by cellular stresses such as telomere shortening and leads to the release of pro-inflammatory cytokines such as interleukin (IL)-6, IL-1β, and IL-1α[9]. The senescence-associated secretory phenotype (SASP) is known to be important for the clearance of senescent cells. However, aging of the immune system can impair its efficiency in clearing senescent cells, exacerbating the pro-inflammatory state[8]. Age-related changes in the innate immune system can reduce phagocyte capability, respiratory burst, and function of natural killer (NK) and dendritic cells, thus impairing immune cell functions and their interactions[8]. Increased production of ROS, generated by the aging process, overwhelms the scavenger systems and leads to the production of damage-associated molecular patterns (DAMPs), activation of the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) (cGAS-STING) pathway, and stimulation of the NLRP3 inflammasome. This results in an extensive inflammatory response. Moreover, telomere shortening is associated with mitochondrial dysfunction due to decreased expression of peroxisome proliferator-activated receptor coactivator-1α and -1β. All of these age-related pathophysiological changes collectively lead to a chronic low-inflammatory state that affects both the innate and adaptive immune systems[25].
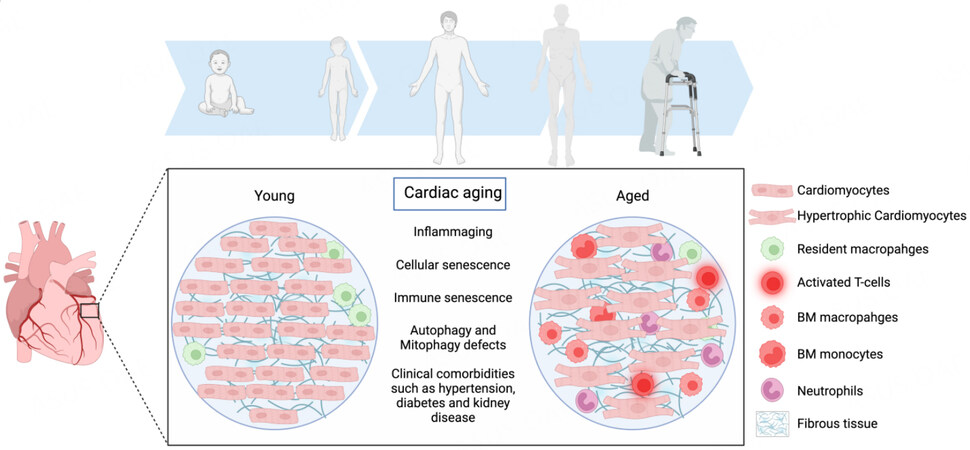
Figure 1. Cardiac aging is associated with a myriad of cellular and structural changes that result in heightened systemic and cardiac inflammation. These changes cause a shift in cardiac macrophage populations with an abundance of bone marrow-derived, pro-inflammatory macrophages and a reduction in the percentage of resident anti-inflammatory macrophages. The heart also becomes populated with other immune cells that promote tissue inflammation, such as monocytes, neutrophils, and T cells. This phenomenon is associated with cardiac fibroblast activation and fibrosis. Additionally, aging is associated with cardiomyocyte hypertrophy and increased stiffness. Taken together, aging-related changes in the heart result in diastolic dysfunction and the development of heart failure with preserved ejection fraction (the figure was prepared using Biorender, https://biorender.com/).
AGE-ASSOCIATED IMMUNE CELL CHANGES AND THEIR EFFECTS ON THE HEART
Aging is associated with a dysregulated innate immunity in both animals and humans. The hallmarks of these phenotypic changes are a heightened baseline inflammatory state and an impaired immune response to external pathogens and stimuli. Mechanistically, this phenomenon is linked to altered myeloid cell production and activation, impaired expression, and activation of PRRs. Collectively, these factors result in a dysregulated immune system, increased cytokine production, and an increased inflammatory state with aging[26].
Significant reduction in the proliferative capacity of hematopoietic progenitor cells in the bone marrow results in the skewed composition of the myeloid cell compartment[27]. In addition to changes in the number of circulating and tissue-bound myeloid cells, studies have demonstrated functional impairment with aging, including reduced chemotaxis, migration, and phagocytosis, as seen among aging neutrophils[28]. Furthermore, a dysregulated neutrophil response to stimuli and reduction in the production of anti-inflammatory and anti-apoptotic cytokines exacerbate the baseline inflammatory state[29]. These changes also extend to natural killer (NK) cells, monocytes, and macrophages during aging. NK cells show a reduction in both cytotoxic and secretory functions with aging, resulting in impaired response to pathogens and dysregulation of immune signaling with other immune cells[30]. Similarly, monocytes and macrophages demonstrate a reduced response to TLRs and cytokine signaling with aging concomitant with inappropriately increased cytokine production[31,32]. Macrophages play a significant role in the development of HFpEF, as they can produce and secrete various pro-inflammatory and pro-fibrotic cytokines, which can lead to the remodeling of the left ventricle[33]. This remodeling can lead to adverse changes in the heart’s contractile and diastolic functions, which can ultimately lead to various forms of heart failure. Additionally, dysregulated macrophages can contribute to other processes that are known to contribute to HFpEF, such as oxidative stress, fibrosis, and vascular stiffening[34]. Therefore, pro-inflammatory macrophages may be an important target for therapeutic interventions in HFpEF. Taken together, aging is associated with dysregulated innate immune system, impaired response to external stimuli, and exacerbated cytokine production resulting in heightened baseline inflammation.
While macrophages are believed to be the predominant immune cell population in the heart during physiological conditions, recent studies demonstrate that additional leucocyte populations populate the heart in the absence of injury. Among these populations, T cells demonstrate significant dynamic changes with aging[35]. Structural changes seen with aging have been correlated with the accumulation of activated CD4+/Foxp3-/IFNγ+ T cells in the mediastinal lymph nodes draining the heart. The shift in T cell populations contributes to age-related cardiac inflammation and dysfunction. Indeed, adoptive transplantation studies of mediastinal lymph node T cells from aged, but not young, mice demonstrated primed activation and cardiotropism, resulting in a mild form of cardiac dysfunction[35]. This study suggests that the contribution of T cells to aging-related cardiac dysfunction is a result of direct effects as well as indirect effects through the regulation of the immune system. The mechanisms behind priming T cells during the cardiac aging process are not fully understood but could be related to exposure to cardiomyocyte components that serve as auto-antigens, which results in an autoreactive immune response[35,36]. In fact, studies have confirmed the role of MHCII+ resident macrophages in presenting CM components to T cells resulting in priming them and a consistent population of T- and B-cells in the heart in the absence of injury[37]. Aging also affects the adaptive immune system through reduced production of naïve T cells and Treg cells, resulting in an imbalance towards a higher percentage of self-reacting T cells with an increased incidence of auto-immune diseases[36]. Similarly, reduction in B-cells, especially those expressing CD28, results in an imbalance between regulatory and pro-inflammatory T cells[38,39]. Collectively, changes in the innate and adaptive immune systems potentiate chronic inflammation in the heart with aging and subsequently contribute to functional deterioration.
HEART FAILURE WITH PRESERVED EJECTION (HFpEF) IN THE AGING POPULATION
The prevalence of HFpEF has risen in the population over the last two decades. This could be attributed to the increasing prevalence of its risk factors, such as aging, obesity, hypertension, and type 2 diabetes mellitus (T2D). In particular, aging is associated with tissue fibrosis, a process that is linked to activated cardiac fibroblasts (CFs), neurohormonal activation, and heightened inflammation [Figure 2]. Multiple age-related cellular pathways that result in positive oxidative stress led to increased ECM production and significant impairment of its clearance, thus tipping the proteolytic balance towards net ECM accumulation, cross-linking, and increased myocardial stiffness. The Angiotensin II (Ang II) system is a key neurohormonal pathway activated in aging that has a significant role in cardiac inflammation through direct and indirect pathways. Ang II binds to angiotensin 1 receptors (AT1) and directly activates cardiac fibroblasts resulting in increased production of extracellular matrix components, collagen deposition in the extracellular space, and cardiac fibrosis. Beyond its direct effects on CFs, Ang II-AT1 signaling promotes inflammation through increased intracellular ROS, mitochondrial dysfunction, and the release of mtDNA. The combination of the direct and indirect effects of AngII-AT1 signaling in aging results in diastolic dysfunction and a higher prevalence of HFpEF[40].
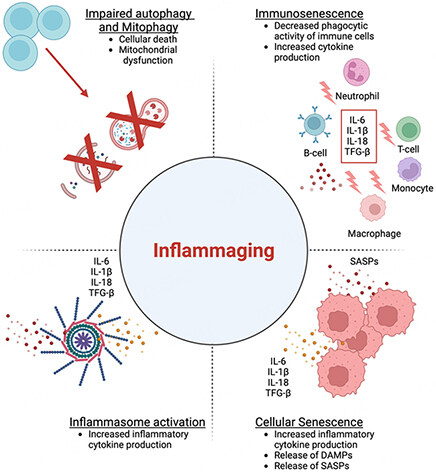
Figure 2. The aging process has a complex impact on the immune system, leading to sustained low-grade immune activation, reduced sensitivity to immunogenic stimuli, and accumulation of senescent cells. These changes result in a chronic inflammatory process known as SASP (senescence-associated secretory phenotype). In addition, increased inflammation with aging may be attributed to inflammasome activation, due to defects in autophagy and mitophagy. This pro-inflammatory milieu perpetuates the chronic low-grade inflammation commonly seen in aging. This figure was modified from Puspitasari et al.[69]. Additionally, the figure was prepared using Biorender, https://biorender.com/).
In addition to HFpEF due to increased cardiac fibrosis, structural cardiac changes can be caused by infiltrating cardiac disease. Autopsy studies have demonstrated a high prevalence of amyloidosis in older people, with the acquired wild-type variant (ATTRwt) being the most common aging-related variant[41]. Mechanistically, aging could destabilize transthyretin through alterations in post-transcriptional changes in the protein or its chaperones[42,43]. Amyloidosis is caused by the deposition of amyloid fibrils in the ECM, resulting in increased wall thickness, increased stiffness, and reduction of contractile function in the long term.
The effects of structural cardiac changes leading to diastolic and systolic dysfunction are exacerbated by the high prevalence of comorbidities in the aging population. These comorbidities profoundly affect cardiac function and structure and include a sedentary lifestyle, hypertension, diabetes, and atrial fibrillation. Among the common comorbidities seen in aging, ischemic heart disease represents a major cause of structural heart disease in older people. Studies have documented a prevalence of obstructive heart disease as high as 60% in individuals over the age of 60[44]. Furthermore, studies have documented poor healing response after myocardial infarction in older people with worse adverse cardiac remodeling and rapid progression to systolic heart failure[45]. In addition, obesity is also prevalent in older people in the US and contributes to the heightened inflammatory state, renin-angiotensin system activation, increased Ang II production, and the development of diastolic heart failure[34,46].
THERAPEUTIC AND TRANSLATIONAL PERSPECTIVES IN CARDIAC INFLAMMAGING
Mitochondrial dysfunction and reduction in AMPK signaling play a critical role in inflammaging through the release of ROS and mtDNA, which activates the innate immune system and contributes to the chronic inflammatory response. Therefore, multiple studies have explored the therapeutic potential of drugs that maintain mitochondrial function during aging. Metformin reduces oxidative stress through the inhibition of mitochondrial complex I, resulting in the activation of AMPK and improved mitochondrial function[47]. Nutraceuticals, such as resveratrol, attenuate mitochondrial-induced inflammaging and endothelial cell dysfunction by activating cAMP-AMPK[48]. Given the detrimental impact of oxidative stress on mitochondrial function, antioxidants such as Coenzyme Q have been proposed as therapeutic alternatives. Clinical trials have shown modest benefits for such approaches in chronic heart failure patients, which could be explained, at least in part, by the lack of specificity to mitochondria[49]. Recent approaches that utilize triphenylphosphonium as a mitochondria-targeted vehicle are being tested in clinical trials[50]. Given the role of senescence in the age-related inflammatory response, senolytic therapies have been proposed as a therapeutic target. Dasatinib, a tyrosine kinase inhibitor, combined with Quercetin, a natural flavonoid with senolytic activity, reduces the effect of aging on vascular function and specifically reduces vascular calcification[51]. Similarly, Navitoclax, which inhibits the apoptosis regulator protein B-cell lymphoma 2
Inflammation plays a critical role in the pathophysiology of cardiac dysfunction with aging. Accordingly, targeting inflammation represents a promising new strategy for the management of age-related cardiovascular diseases. Among the various pathways targeted in clinical studies, the inhibition of NLRP3 inflammasome and the downstream IL-1β signaling appear to be the most effective targets. Colchicine, which inhibits tubulin polymerization and the assembly of the multimeric NLRP3 inflammasome, reduces major cardiovascular events (MACE) in patients with chronic stable coronary artery disease[53,54] as well as those with recent myocardial infarction[55]. However, colchicine did not improve clinical outcomes when administered immediately after myocardial infarction suggesting patient- and clinical-scenario-specific benefits. NLRP3 activation results in the release and activation of IL-1β and IL-6, major pro-inflammatory cytokines.
The CANTOS trial, a landmark randomized controlled clinical trial, found that canakinumab, a monoclonal antibody directed against interleukin-1β, significantly reduced the risk of MACE in patients with a history of myocardial infarction and elevated levels of high-sensitivity C-reactive protein (hs-CRP). The trial included 10,061 participants from 33 countries with a median follow-up of 3.7 years. It demonstrated a 15% reduction in all-cause mortality, a 16% reduction in MACE, and a 25% reduction in inflammatory markers. Additionally, canakinumab reduced hs-CRP levels and the risk of all-cause mortality among patients with elevated CRP[56]. Similar findings were observed among patients with recent myocardial infarction[57,58]. In the VCU-ART pilot studies, forty patients with STEMI who underwent fourteen days of anakinra treatment showed a reduction in the area under the curve for hs-CRP, and a signal for reduced progression to HF when compared to placebo[59-61]. The MRC-ILA-Heart study of 182 patients with non-ST-segment elevation myocardial infarction (NSTEMI) revealed that IL-1 blockade with anakinra successfully reduced CRP levels at 7 days post-NSTEMI yet failed to yield any improvement in clinical outcomes[62]. Given that the majority of benefits from Canakinumab is in patients with documented inflammation and high risk of cardiovascular events, some experts suggest that canakinumab may be considered for patients with prior myocardial infarction, high-sensitivity C-reactive protein levels of 2 mg/L or greater, and low-density lipoprotein cholesterol levels below 70 mg/dL, who are at high risk of recurrent events despite optimal medical therapy. However, this recommendation is not widely accepted, and more evidence of cost-effectiveness is needed to support it due to the high cost of Canakinumab.
Other targetable cytokines in the systemic inflammatory response include IL-6. The RESCUE trial was a phase 2 trial that enrolled 264 patients with moderate to severe chronic kidney disease and high-sensitivity CRP of at least 2 mg/L[2]. Patients were randomly assigned to receive a placebo or ziltivekimab, an IL-6 ligand monoclonal antibody, at 7.5 mg, 15 mg, or 30 mg every 4 weeks up to 24 weeks[63]. The primary endpoint was a change in high-sensitivity CRP from baseline to week 12. The secondary endpoints included changes in other inflammatory and thrombotic biomarkers, such as fibrinogen, D-dimer, IL-6, and sgp130. In this study, ziltivekimab significantly reduced multiple biomarkers of systemic inflammation and the magnitude of change in high-sensitivity CRP with ziltivekimab was nearly twice as large in RESCUE as in the recent CANTOS trial of canakinumab. The ZEUS study is a phase 3 trial that is currently enrolling patients with chronic kidney disease and elevated high-sensitivity CRP to test whether ziltivekimab can reduce cardiovascular events. The primary endpoint is a composite of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke and the results are expected in 2024 (https://clinicaltrials.gov/).
A word of caution is necessary when targeting inflammation, particularly in older people with a dysregulated immune system. Many of these therapies result in an increased risk of infections and, in the case of colchicine, increased non-cardiac death. This highlights the need for more targeted immunomodulatory therapies with fewer immunosuppressive effects.
Multiple nonpharmacological approaches have been adopted to mitigate the effects of aging on the cardiovascular system. Nonpharmacological approaches using CAR T cells that target senescent cells are demonstrating promise in pre-clinical and early clinical studies[64]. Recent studies have demonstrated that regular exercise can help reduce inflammation associated with aging[65]. Physical activity was associated with lower circulating markers of inflammation such as pro-inflammatory cytokines (IL-1β and IL-18), and this phenomenon is mediated through the methylation of the apoptosis-associated protein caspase gene (ASC) and the downstream NLRP3 inflammasome[66]. Regular exercise has been shown to reduce inflammation, with evidence from epidemiological studies and randomized control trials demonstrating inverse associations between physical activity and markers of low-grade systemic inflammation[67]. Exercise activates molecular signals that may circumvent defects in insulin signaling in the skeletal muscle and increases the mitochondria of skeletal muscle, which is associated with an improvement of the insulin sensitivity in the skeletal muscle, thus improving the age-related effects of T2D[68]. Additionally, exercise halts telomere shortening, mitochondrial dysfunction, cellular senescence, and the production of SASP factors[67].
CONCLUSION
The number of older people continues to increase steadily. Consequently, there is an urgent need for therapies that can combat the effects of aging on the cardiovascular system. New insights into the mechanism of aging-related cellular dysfunction have paved the way for new therapies with an evolving body of basic research evidence. Ongoing and future translational studies are expected to demonstrate clinical evidence for the efficacy of new therapeutic strategies that mitigate age-related cardiac dysfunction. In addition to new pharmacological strategies, lifestyle modifications and exercise have been shown to modulate the cellular effects of aging and reduce the magnitude of inflammaging and associated cardiovascular disease. Current evidence suggests that age-related cardiovascular effects could become a treatable condition in the future.
DECLARATIONS
Authors’ contributionsConceived and wrote the paper: Goldstein DR, Abdel-Latif A
Availability of data and materialsNot applicable.
Financial support and sponsorshipDr. Abdel-Latif is supported by NIH Grant (R01 HL138488). Dr. Daniel R. Goldstein is supported by NIH grants (AG028082, HL155169 and AI138347).
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Cheng S, Fernandes VR, Bluemke DA, McClelland RL, Kronmal RA, Lima JA. Age-related left ventricular remodeling and associated risk for cardiovascular outcomes: the Multi-Ethnic Study of Atherosclerosis. Circ Cardiovasc Imaging 2009;2:191-8.
2. Nayor M, Cooper LL, Enserro DM, et al. Left ventricular diastolic dysfunction in the community: impact of diagnostic criteria on the burden, correlates, and prognosis. J Am Heart Assoc 2018;7:e008291.
3. Christou DD, Seals DR. Decreased maximal heart rate with aging is related to reduced {beta}-adrenergic responsiveness but is largely explained by a reduction in intrinsic heart rate. J Appl Physiol 2008;105:24-9.
4. English BA, Appalsamy M, Diedrich A, et al. Tachycardia, reduced vagal capacity, and age-dependent ventricular dysfunction arising from diminished expression of the presynaptic choline transporter. Am J Physiol Heart Circ Physiol 2010;299:H799-810.
5. Yan L, Vatner DE, O'Connor JP, et al. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell 2007;130:247-58.
6. Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol 2008;51:802-9.
7. Jalife J, Kaur K. Atrial remodeling, fibrosis, and atrial fibrillation. Trends Cardiovasc Med 2015;25:475-84.
8. Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol 2018;14:576-90.
9. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013;153:1194-217.
10. Kundu P, Blacher E, Elinav E, Pettersson S. Our gut microbiome: the evolving inner self. Cell 2017;171:1481-93.
11. Lee YK, Mazmanian SK. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 2010;330:1768-73.
12. Libby P, Sidlow R, Lin AE, et al. Clonal hematopoiesis: crossroads of aging, cardiovascular disease, and cancer: JACC review topic of the week. J Am Coll Cardiol 2019;74:567-77.
13. de Arellano ML, Pozdniakova S, Kühl AA, Baczko I, Ladilov Y, Regitz-Zagrosek V. Sex differences in the aging human heart: decreased sirtuins, pro-inflammatory shift and reduced anti-oxidative defense. Aging 2019;11:1918-33.
14. Barcena ML, Pozdniakova S, Haritonow N, et al. Dilated cardiomyopathy impairs mitochondrial biogenesis and promotes inflammation in an age- and sex-dependent manner. Aging 2020;12:24117-33.
15. Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 2004;23:2369-80.
16. Tyrrell DJ, Blin MG, Song J, Wood SC, Goldstein DR. Aging impairs mitochondrial function and mitophagy and elevates interleukin 6 within the cerebral vasculature. J Am Heart Assoc 2020;9:e017820.
17. Tyrrell DJ, Blin MG, Song J, et al. Age-associated mitochondrial dysfunction accelerates atherogenesis. Circ Res 2020;126:298-314.
18. Tyrrell DJ, Goldstein DR. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat Rev Cardiol 2021;18:58-68.
19. Salminen A, Kaarniranta K, Kauppinen A. Age-related changes in AMPK activation: Role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling pathways. Ageing Res Rev 2016;28:15-26.
20. Sundaresan NR, Vasudevan P, Zhong L, et al. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat Med 2012;18:1643-50.
21. Zeng H, Chen JX. Sirtuin 3, Endothelial metabolic reprogramming, and heart failure with preserved ejection fraction. J Cardiovasc Pharmacol 2019;74:315-23.
22. Pålsson-McDermott EM, O'Neill LAJ. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res 2020;30:300-14.
23. Soto-Heredero G, Gómez de Las Heras MM, Gabandé-Rodríguez E, Oller J, Mittelbrunn M. Glycolysis - a key player in the inflammatory response. FEBS J 2020;287:3350-69.
24. Bess E, Fisslthaler B, Frömel T, Fleming I. Nitric oxide-induced activation of the AMP-activated protein kinase α2 subunit attenuates IκB kinase activity and inflammatory responses in endothelial cells. PLoS One 2011;6:e20848.
25. Sahin E, Colla S, Liesa M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011;470:359-65.
26. Panda A, Qian F, Mohanty S, et al. Age-associated decrease in TLR function in primary human dendritic cells predicts influenza vaccine response. J Immunol 2010;184:2518-27.
27. Dykstra B, Olthof S, Schreuder J, Ritsema M, de Haan G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med 2011;208:2691-703.
28. Butcher SK, Chahal H, Nayak L, et al. Senescence in innate immune responses: reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J Leukoc Biol 2001;70:881-6.
29. Tortorella C, Simone O, Piazzolla G, Stella I, Cappiello V, Antonaci S. Role of phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways in granulocyte macrophage-colony-stimulating factor failure to delay fas-induced neutrophil apoptosis in elderly humans. J Gerontol A Biol Sci Med Sci 2006;61:1111-8.
30. Nogusa S, Ritz BW, Kassim SH, Jennings SR, Gardner EM. Characterization of age-related changes in natural killer cells during primary influenza infection in mice. Mech Ageing Dev 2008;129:223-30.
31. Kissin E, Tomasi M, McCartney-Francis N, Gibbs CL, Smith PD. Age-related decline in murine macrophage production of nitric oxide. J Infect Dis 1997;175:1004-7.
32. Tasat DR, Mancuso R, O'Connor S, Molinari B. Age-dependent change in reactive oxygen species and nitric oxide generation by rat alveolar macrophages. Aging Cell 2003;2:159-64.
33. Pinto AR, Godwin JW, Chandran A, et al. Age-related changes in tissue macrophages precede cardiac functional impairment. Aging 2014;6:399-413.
34. Shaw AC, Goldstein DR, Montgomery RR. Age-dependent dysregulation of innate immunity. Nat Rev Immunol 2013;13:875-87.
35. Ramos GC, van den Berg A, Nunes-Silva V, et al. Myocardial aging as a T-cell-mediated phenomenon. Proc Natl Acad Sci USA 2017;114:E2420-9.
36. Thiault N, Darrigues J, Adoue V, et al. Peripheral regulatory T lymphocytes recirculating to the thymus suppress the development of their precursors. Nat Immunol 2015;16:628-34.
37. Haghikia A, Kaya Z, Schwab J, et al. Evidence of autoantibodies against cardiac troponin I and sarcomeric myosin in peripartum cardiomyopathy. Basic Res Cardiol 2015;110:60.
38. Pangrazzi L, Reidla J, Carmona Arana JA, et al. CD28 and CD57 define four populations with distinct phenotypic properties within human CD8+ T cells. Eur J Immunol 2020;50:363-79.
40. Cieslik KA, Taffet GE, Carlson S, Hermosillo J, Trial J, Entman ML. Immune-inflammatory dysregulation modulates the incidence of progressive fibrosis and diastolic stiffness in the aging heart. J Mol Cell Cardiol 2011;50:248-56.
41. Tanskanen M, Peuralinna T, Polvikoski T, et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med 2008;40:232-9.
42. Buxbaum JN, Tagoe C, Gallo G, Walker JR, Kurian S, Salomon DR. Why are some amyloidoses systemic? FASEB J 2012;26:2283-93.
43. Zhao L, Buxbaum JN, Reixach N. Age-related oxidative modifications of transthyretin modulate its amyloidogenicity. Biochemistry 2013;52:1913-26.
44. Roger VL, Weston SA, Killian JM, et al. Time trends in the prevalence of atherosclerosis: a population-based autopsy study. Am J Med 2001;110:267-73.
45. Gaudron P, Eilles C, Kugler I, Ertl G. Progressive left ventricular dysfunction and remodeling after myocardial infarction. Potential mechanisms and early predictors. Circulation 1993;87:755-63.
46. Liberale L, Montecucco F, Tardif JC, Libby P, Camici GG. Inflamm-ageing: the role of inflammation in age-dependent cardiovascular disease. Eur Heart J 2020;41:2974-82.
47. Kulkarni AS, Gubbi S, Barzilai N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab 2020;32:15-30.
48. Gupta SC, Kunnumakkara AB, Aggarwal S, Aggarwal BB. Inflammation, a double-edge sword for cancer and other age-related diseases. Front Immunol 2018;9:2160.
49. Mortensen SA, Rosenfeldt F, Kumar A, et al. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: results from Q-SYMBIO: a randomized double-blind trial. JACC Heart Fail 2014;2:641-9.
50. Forini F, Canale P, Nicolini G, Iervasi G. Mitochondria-targeted drug delivery in cardiovascular disease: a long road to nano-cardio medicine. Pharmaceutics 2020;12:1122.
51. Roos CM, Zhang B, Palmer AK, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016;15:973-7.
52. Walaszczyk A, Dookun E, Redgrave R, et al. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 2019;18:e12945.
53. Nidorf SM, Fiolet ATL, Mosterd A, et al. Colchicine in patients with chronic coronary disease. N Engl J Med 2020;383:1838-47.
54. Nidorf SM, Eikelboom JW, Budgeon CA, Thompson PL. Low-dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol 2013;61:404-10.
55. Tardif JC, Kouz S, Waters DD, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med 2019;381:2497-505.
56. Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017;377:1119-31.
57. Buckley LF, Carbone S, Trankle CR, et al. Effect of interleukin-1 blockade on left ventricular systolic performance and work: a post hoc pooled analysis of 2 clinical trials. J Cardiovasc Pharmacol 2018;72:68-70.
58. Buckley LF, Abbate A. Interleukin-1 blockade in cardiovascular diseases: a clinical update. Eur Heart J 2018;39:2063-9.
59. Abbate A, Kontos MC, Abouzaki NA, et al. Comparative safety of interleukin-1 blockade with anakinra in patients with ST-segment elevation acute myocardial infarction (from the VCU-ART and VCU-ART2 pilot studies). Am J Cardiol 2015;115:288-92.
60. Abbate A, Kontos MC, Grizzard JD, et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study). Am J Cardiol 2010;105:1371-7.e1.
61. Abbate A, Van Tassell BW, Biondi-Zoccai G, et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study]. Am J Cardiol 2013;111:1394-400.
62. Morton AC, Rothman AM, Greenwood JP, et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: the MRC-ILA Heart Study. Eur Heart J 2015;36:377-84.
63. Ridker PM, Devalaraja M, Baeres FMM, et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021;397:2060-9.
64. Feucht J, Abou-El-Enein M. Senolytic CAR T cells in solid tumors and age-related pathologies. Mol Ther 2020;28:2108-10.
65. Hamer M. The relative influences of fitness and fatness on inflammatory factors. Prev Med 2007;44:3-11.
66. Nakajima K, Takeoka M, Mori M, et al. Exercise effects on methylation of ASC gene. Int J Sports Med 2010;31:671-5.
67. Li H, Hastings MH, Rhee J, Trager LE, Roh JD, Rosenzweig A. Targeting age-related pathways in heart failure. Circ Res 2020;126:533-51.
68. Carapeto PV, Aguayo-Mazzucato C. Effects of exercise on cellular and tissue aging. Aging 2021;13:14522-43.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Goldstein DR, Abdel-Latif A. Immune mechanisms of cardiac aging. J Cardiovasc Aging 2023;3:17. http://dx.doi.org/10.20517/jca.2023.02
AMA Style
Goldstein DR, Abdel-Latif A. Immune mechanisms of cardiac aging. The Journal of Cardiovascular Aging. 2023; 3(2): 17. http://dx.doi.org/10.20517/jca.2023.02
Chicago/Turabian Style
Goldstein, Daniel R., Ahmed Abdel-Latif. 2023. "Immune mechanisms of cardiac aging" The Journal of Cardiovascular Aging. 3, no.2: 17. http://dx.doi.org/10.20517/jca.2023.02
ACS Style
Goldstein, DR.; Abdel-Latif A. Immune mechanisms of cardiac aging. J. Cardiovasc. Aging. 2023, 3, 17. http://dx.doi.org/10.20517/jca.2023.02
About This Article
Copyright

Data & Comments
Data


 Cite This Article 13 clicks
Cite This Article 13 clicks

 Like This Article 24
likes
Like This Article 24
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.